OBJECTIVES
After studying this chapter, you should be able to:
Describe the components of blood and lymph, their origins, and the role of hemoglobin in transporting oxygen in red blood cells.
Understand the molecular basis of blood groups and the reasons for transfusion reactions.
Delineate the process of hemostasis that restricts blood loss when vessels are damaged, and the adverse consequences of intravascular thrombosis.
Identify the types of blood and lymphatic vessels that make up the circulatory system and the regulation and function of their primary constituent cell types.
Describe how physical principles dictate the flow of blood and lymph around the body.
Understand the basis of methods used to measure blood flow and blood pressure in various vascular segments.
Understand the basis of disease states where components of the blood and vasculature are abnormal, dysregulated, or both.
INTRODUCTION
The circulatory system supplies inspired O2 as well as substances absorbed from the gastrointestinal tract to the tissues, returns CO2 to the lungs and other products of metabolism to the kidneys, functions in the regulation of body temperature, and distributes hormones and other agents that regulate cell function. The blood, the carrier of these substances, is pumped through a closed system of blood vessels by the heart. From the left ventricle, blood is pumped through the arteries and arterioles to the capillaries, where it equilibrates with the interstitial fluid. The capillaries drain through venules into the veins and back to the right atrium. Some tissue fluids enter another system of closed vessels, the lymphatics, which drain lymph via the thoracic duct and the right lymphatic duct into the venous system. The circulation is controlled by multiple regulatory systems that function in general to maintain adequate capillary blood flow when possible in all organs, but particularly in the heart and brain.
Blood flows through the circulation primarily because of the forward motion imparted to it by the pumping of the heart, although in the case of the systemic circulation, diastolic recoil of the walls of the arteries, compression of the veins by skeletal muscles during exercise, and the negative pressure in the thorax during inspiration also move the blood forward. The resistance to flow depends to a minor degree on the viscosity of the blood but mostly on the diameter of the vessels, and principally that of the arterioles. The blood flow to each tissue is regulated by local chemical and general neural and humoral mechanisms that dilate or constrict its vessels. All of the blood flows through the lungs, but the systemic circulation is made up of numerous different circuits in parallel (Figure 31–1). The arrangement permits wide variations in regional blood flow without changing total systemic flow.

This chapter is concerned with blood and lymph and with the multiple functions of the cells they contain. It will also address general principles that apply to all parts of the circulation and pressure and flow in the systemic circulation. The homeostatic mechanisms operating to adjust flow are the subject of Chapter 32. The special characteristics of pulmonary and renal circulation are discussed in Chapters 34 and 37. Likewise, the role of blood as the carrier of many immune effector cells will not be discussed here, but rather was covered in Chapter 3.
BLOOD AS A CIRCULATORY FLUID
BONE MARROW
In the adult, red blood cells, many white blood cells, and platelets are formed in the bone marrow. In the fetus, blood cells are also formed in the liver and spleen, and in adults such extramedullary hematopoiesis may occur in diseases in which the bone marrow becomes destroyed or fibrosed. In children, blood cells are actively produced in the marrow cavities of all the bones. By age 20, the marrow in the cavities of the long bones, except for the upper humerus and femur, has become inactive (Figure 31–2). Active cellular marrow is called red marrow; inactive marrow that is infiltrated with fat is called yellow marrow.

The bone marrow is actually one of the largest organs in the body, approaching the size and weight of the liver. It is also one of the most active. Normally, 75% of the cells in the marrow belong to the white blood cell–producing myeloid series and only 25% are maturing red cells, even though there are over 500 times as many red cells in the circulation as there are white cells. This difference in the marrow reflects the fact that the average life span of white cells is short, whereas that of red cells is long.
Hematopoietic stem cells (HSCs) are bone marrow cells that are capable of producing all types of blood cells. They differentiate into one or another type of committed stem cells (progenitor cells). These in turn form the various differentiated types of blood cells. There are separate pools of progenitor cells for megakaryocytes, lymphocytes, erythrocytes, eosinophils, and basophils; neutrophils and monocytes arise from a common precursor. The bone marrow stem cells are also the source of osteoclasts (see Chapter 21), Kupffer cells (see Chapter 28), mast cells, dendritic cells, and Langerhans cells. The HSCs are few in number but are capable of completely replacing the bone marrow when injected into a patient whose own bone marrow has been entirely destroyed.
The HSCs are derived from uncommitted, totipotent stem cells that can be stimulated to form any cell in the body. Adults have a few of these, but they are more readily obtained from the blastocysts of embryos. There is not surprisingly immense interest in stem cell research due to its potential to regenerate diseased tissues, but ethical issues are involved, and debate on these issues will undoubtedly continue.
WHITE BLOOD CELLS
Normally, human blood contains 4000–11,000 white blood cells per microliter (Table 31–1). Of these, the granulocytes (polymorphonuclear leukocytes, PMNs) are the most numerous. Young granulocytes have horseshoe-shaped nuclei that become multilobed as the cells grow older (Figure 31–3). Most of them contain neutrophilic granules (neutrophils), but a few contain granules that stain with acidic dyes (eosinophils), and some have basophilic granules (basophils). The other two cell types found normally in peripheral blood are lymphocytes, which have large round nuclei and scanty cytoplasm, and monocytes, which have abundant agranular cytoplasm and kidney-shaped nuclei (Figure 31–3). Acting together, these cells provide the body with the powerful defenses against tumors and viral, bacterial, and parasitic infections that were discussed in Chapter 3.
FIGURE 31–3
Development of various formed elements of the blood from bone marrow cells. Cells below the horizontal line are found in normal peripheral blood. The principal sites of action of erythropoietin (erythro) and the various colony-stimulating factors (CSF) that stimulate the differentiation of the components are indicated. G, granulocyte; M, macrophage; IL, interleukin; thrombo, thrombopoietin; erythro, erythropoietin; SCF, stem cell factor.

| Cell | Cells/μL (average) | Approximate Normal Range | Percentage of Total White Cells |
|---|---|---|---|
| Total white blood cells | 9000 | 4000–11,000 | … |
| Granulocytes | |||
| Neutrophils | 5400 | 3000–6000 | 50–70 |
| Eosinophils | 275 | 150–300 | 1–4 |
| Basophils | 35 | 0–100 | 0.4 |
| Lymphocytes | 2750 | 1500–4000 | 20–40 |
| Monocytes | 540 | 300–600 | 2–8 |
| Erythrocytes | |||
| Females | 4.8 × 106 | … | … |
| Males | 5.4 × 106 | … | … |
| Platelets | 300,000 | 200,000–500,000 | … |
PLATELETS
Platelets are small, granulated bodies that aggregate at sites of vascular injury. They lack nuclei and are 2–4 μm in diameter (Figure 31–3). There are about 300,000/μL of circulating blood, and they normally have a half-life of about 4 days. The megakaryocytes, giant cells in the bone marrow, form platelets by pinching off bits of cytoplasm and extruding them into the circulation. Between 60% and 75% of the platelets that have been extruded from the bone marrow are in the circulating blood, and the remainder are mostly in the spleen. Splenectomy causes an increase in the platelet count (thrombocytosis).
RED BLOOD CELLS
The red blood cells (erythrocytes) carry hemoglobin in the circulation. They are biconcave disks (Figure 31–4) that are manufactured in the bone marrow. In mammals, they lose their nuclei before entering the circulation. In humans, they survive in the circulation for an average of 120 days. The average normal red blood cell count is 5.4 million/μL in men and 4.8 million/μL in women. The number of red cells is also conveniently expressed as the hematocrit, or the percentage of the blood, by volume, that is occupied by erythrocytes. Each human red blood cell is about 7.5 μm in diameter and 2 μm thick, and each contains approximately 29 pg of hemoglobin (Table 31–2). There are thus about 3 × 1013 red blood cells and about 900 g of hemoglobin in the circulating blood of an adult man (Figure 31–5).
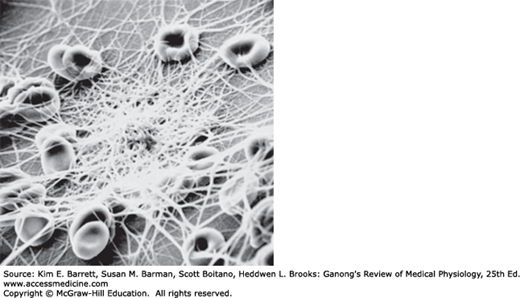

| Male | Female | ||
|---|---|---|---|
| Hematocrit (Hct) (%) | 47 | 42 | |
| Red blood cells (RBC) (106/μL) | 5.4 | 4.8 | |
| Hemoglobin (Hb) (g/dL) | 16 | 14 | |
| Mean corpuscular volume (MCV) (fL) | 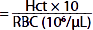 | 87 | 87 |
| Mean corpuscular hemoglobin (MCH) (pg) | 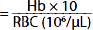 | 29 | 29 |
| Mean corpuscular hemoglobin concentration (MCHC) (g/dL) | 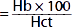 | 34 | 34 |
| Mean cell diameter (MCD) (μm) | = Mean diameter of 500 cells in smear | 7.5 | 7.5 |
The feedback control of erythropoiesis by erythropoietin is discussed in Chapter 38, and the role of IL-1, IL-3, IL-6 (interleukin), and GM-CSF (granulocyte-macrophage colony-stimulating factor) in development of the relevant erythroid stem cells is shown in Figure 31–3.
The spleen is an important blood filter that removes aged or abnormal red cells. It also contains many platelets and plays a significant role in the immune system. Abnormal red cells are removed if they are not as flexible as normal red cells and consequently are unable to squeeze through the slits between the endothelial cells that line the splenic sinuses (Clinical Box 31–1).
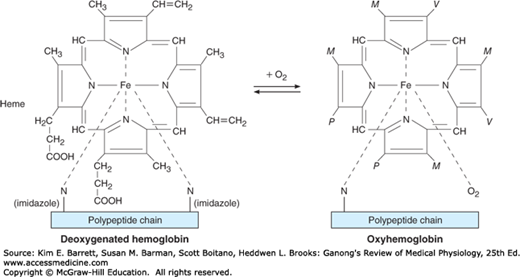
The red, oxygen-carrying pigment in the red blood cells of vertebrates is hemoglobin, a protein with a molecular weight of 64,450. Hemoglobin is a globular molecule made up of four subunits (Figure 31–6). Each subunit contains a heme moiety conjugated to a polypeptide. Heme is an iron-containing porphyrin derivative (Figure 31–7). The polypeptides are referred to collectively as the globin portion of the hemoglobin molecule. There are two pairs of polypeptides in each hemoglobin molecule. In normal adult human hemoglobin (hemoglobin A), the two polypeptides are called α chains and β chains. Thus, hemoglobin A is designated α2β2. Not all the hemoglobin in the blood of normal adults is hemoglobin A. About 2.5% of the hemoglobin is hemoglobin A2, in which β chains are replaced by δ chains (α2δ2). The δ chains contain 10 individual amino acid residues that differ from those in β chains.
FIGURE 31–6
Diagrammatic representation of a molecule of hemoglobin A, showing the four subunits. There are two α and two β polypeptide chains, each containing a heme moiety. These moieties are represented by the blue disks. (Reproduced with permission from Harper HA, et al: Physiologische Chemie. Springer, 1975.)

There are small amounts of hemoglobin A derivatives closely associated with hemoglobin A that represent glycated hemoglobins. One of these, hemoglobin A1c (HbA1c), has a glucose attached to the terminal valine in each β chain and is of special interest because it increases in the blood of patients with poorly controlled diabetes mellitus (see Chapter 24), and is measured clinically as a marker of the progression of that disease and/or the effectiveness of treatment.
O2 binds to the Fe2+ in the heme moiety of hemoglobin to form oxyhemoglobin. The affinity of hemoglobin for O2 is affected by pH, temperature, and the concentration in the red cells of 2,3-bisphosphoglycerate (2,3-BPG). 2,3-BPG and H+ compete with O2 for binding to deoxygenated hemoglobin, decreasing the affinity of hemoglobin for O2 by shifting the positions of the four peptide chains (quaternary structure). The details of the oxygenation and deoxygenation of hemoglobin and the physiologic role of these reactions in O2 transport are discussed in Chapter 35.
When blood is exposed to various drugs and other oxidizing agents in vitro or in vivo, the ferrous iron (Fe2+) that is normally present in hemoglobin is converted to ferric iron (Fe3+), forming methemoglobin. Methemoglobin is dark-colored, and when it is present in large quantities in the circulation, it causes a dusky discoloration of the skin resembling cyanosis (see Chapter 35). Some oxidation of hemoglobin to methemoglobin occurs normally, but an enzyme system in the red cells, the dihydronicotinamide adenine dinucleotide (NADH)-methemoglobin reductase system, converts methemoglobin back to hemoglobin. Congenital absence of this system is one cause of hereditary methemoglobinemia.
Carbon monoxide reacts with hemoglobin to form carbon monoxyhemoglobin (carboxyhemoglobin). The affinity of hemoglobin for O2 is much lower than its affinity for carbon monoxide, which consequently displaces O2 on hemoglobin, reducing the oxygen-carrying capacity of blood (see Chapter 35).
The blood of the human fetus normally contains fetal hemoglobin (hemoglobin F). Its structure is similar to that of hemoglobin A except that the β chains are replaced by γ chains; that is, hemoglobin F is α2γ2. The γ chains have 37 amino acid residues that differ from those in the β chain. Fetal hemoglobin is normally replaced by adult hemoglobin soon after birth (Figure 31–8). In certain individuals, it fails to disappear and persists throughout life. In the body, its O2 content at a given PO2 is greater than that of adult hemoglobin because it binds 2,3-BPG less avidly. Hemoglobin F is therefore critical to facilitate movement of O2 from the maternal to the fetal circulation, particularly at later stages of gestation where oxygen demand increases (see Chapter 33). In young embryos there are, in addition, ζ and ε chains, forming Gower 1 hemoglobin (ζ2ε2) and Gower 2 hemoglobin (α2ε2). Switching from one form of hemoglobin to another during development seems to be regulated largely by oxygen availability, with relative hypoxia favoring the production of hemoglobin F both via direct effects on globin gene expression, as well as upregulated production of erythropoietin.

The average normal hemoglobin content of blood is 16 g/dL in men and 14 g/dL in women, all of it in red cells. In the body of a 70-kg man, there are about 900 g of hemoglobin, and 0.3 g of hemoglobin is destroyed and 0.3 g synthesized every hour (Figure 31–5). The heme portion of the hemoglobin molecule is synthesized from glycine and succinyl-CoA (Clinical Box 31–2).
When old red blood cells are destroyed by tissue macrophages, the globin portion of the hemoglobin molecule is split off, and the heme is converted to biliverdin. The enzyme involved is a subtype of heme oxygenase (see Figure 28–4), and CO is formed in the process. CO is an intercellular messenger, like NO (see Chapters 2 and 3). In humans, most of the biliverdin is converted to bilirubin and excreted in the bile (see Chapter 28). The iron from the heme is reused for hemoglobin synthesis.
CLINICAL BOX 31–1 Red Cell Fragility
Red blood cells, like other cells, shrink in solutions with an osmotic pressure greater than that of normal plasma. In solutions with a lower osmotic pressure they swell, become spherical rather than disk-shaped, and eventually lose their hemoglobin (hemolysis). The hemoglobin of hemolyzed red cells dissolves in the plasma, coloring it red. A 0.9% sodium chloride solution is isotonic with plasma. When osmotic fragility is normal, red cells begin to hemolyze when suspended in 0.5% saline; 50% lysis occurs in 0.40–0.42% saline, and lysis is complete in 0.35% saline. In hereditary spherocytosis (congenital hemolytic icterus), the cells are spherocytic in normal plasma and hemolyze more readily than normal cells in hypotonic sodium chloride solutions. Abnormal spherocytes are also trapped and destroyed in the spleen, meaning that hereditary spherocytosis is one of the most common causes of hereditary hemolytic anemia. The spherocytosis is caused by mutations in proteins that make up the membrane skeleton of the erythrocyte, which normally maintain the shape and flexibility of the red cell membrane, including spectrin, the transmembrane protein band 3, and the linker protein, ankyrin. Red cells can also be lysed by drugs (especially penicillin and sulfa drugs) and infections. The susceptibility of red cells to hemolysis by these agents is increased by deficiency of the enzyme glucose-6-phosphate dehydrogenase (G6PD), which catalyzes the initial step in the oxidation of glucose via the hexose monophosphate pathway (see Chapter 1). This pathway generates dihydronicotinamide adenine dinucleotide phosphate (NADPH), which is needed for the maintenance of normal red cell fragility. Severe G6PD deficiency also inhibits the killing of bacteria by granulocytes and predisposes to severe infections.
THERAPEUTIC HIGHLIGHTSSevere cases of hereditary spherocytosis can be treated by splenectomy, but this is not without other risks, such as sepsis. Milder cases can be treated with dietary folate supplementation and/or blood transfusions. Treatment of other forms of hemolytic anemia depends on the underlying cause. Some forms are autoimmune in nature, and have been shown to benefit from treatment with corticosteroids.
Exposure of the skin to white light converts bilirubin to lumirubin, which has a shorter half-life than bilirubin. Phototherapy (exposure to light) is of value in treating infants with jaundice due to hemolysis. Iron is essential for hemoglobin synthesis; if blood is lost from the body and the iron deficiency is not corrected, iron deficiency anemia results.
BLOOD TYPES
The membranes of human red cells contain a variety of blood group antigens, which are also called agglutinogens. The most important and best known of these are the A and B antigens, but there are many more.
The A and B antigens are inherited as mendelian dominants, and individuals are divided into four major blood types on this basis. Type A individuals have the A antigen, type B have the B, type AB have both, and type O have neither. The A and B antigens are complex oligosaccharides that differ in their terminal sugar. An H gene codes for a fucose transferase that adds a terminal fucose, forming the H antigen that is usually present in individuals of all blood types (Figure 31–9). Individuals who are type A also express a second transferase that catalyzes placement of a terminal N-acetylgalactosamine on the H antigen, whereas individuals who are type B express a transferase that places a terminal galactose. Individuals who are type AB have both transferases. Individuals who are type O have neither, so the H antigen persists.
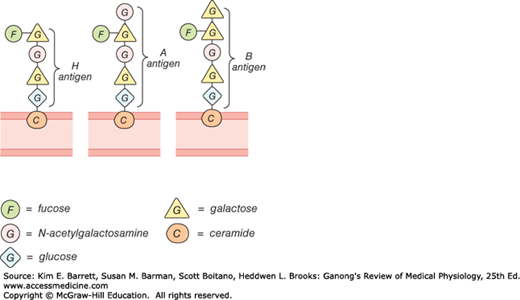
Antibodies against red cell agglutinogens are called agglutinins. Antigens very similar to A and B are common in intestinal bacteria and possibly in foods to which newborn individuals are exposed. Therefore, infants rapidly develop antibodies against the antigens not present in their own cells. Thus, type A individuals develop anti-B antibodies, type B individuals develop anti-A antibodies, type O individuals develop both, and type AB individuals develop neither (Table 31–3). When the plasma of a type A individual is mixed with type B red cells, the anti-B antibodies cause the type B red cells to clump (agglutinate), as shown in Figure 31–10. The other agglutination reactions produced by mismatched plasma and red cells are summarized in Table 31–3. ABO blood typing is performed by mixing an individual’s red blood cells with antisera containing the various agglutinins on a slide and seeing whether agglutination occurs.
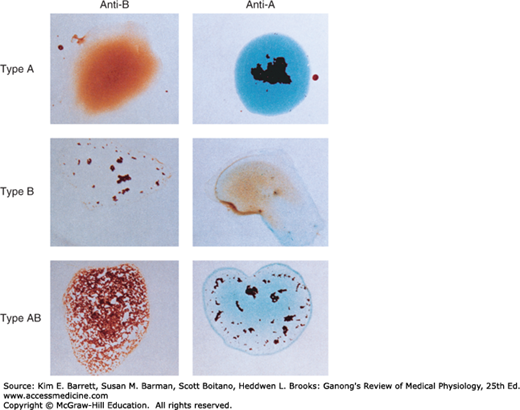
Dangerous hemolytic transfusion reactions occur when blood is transfused into an individual with an incompatible blood type; that is, an individual who has agglutinins against the red cells in the transfusion. The plasma in the transfusion is usually so diluted in the recipient that it rarely causes agglutination even when the titer of agglutinins against the recipient’s cells is high. However, when the recipient’s plasma has agglutinins against the donor’s red cells, the cells agglutinate and hemolyze. Free hemoglobin is liberated into the plasma. The severity of the resulting transfusion reaction may vary from an asymptomatic minor rise in the plasma bilirubin level to severe jaundice and renal tubular damage leading to anuria and death.
CLINICAL BOX 31–2 Abnormalities of Hemoglobin Production
There are two major types of inherited disorders of hemoglobin in humans: the hemoglobinopathies, in which abnormal globin polypeptide chains are produced, and the thalassemias and related disorders, in which the chains are normal in structure but produced in decreased amounts or absent because of defects in the regulatory portion of the globin genes. Mutant genes that cause the production of abnormal hemoglobins are widespread, and over 1000 abnormal hemoglobins have been described in humans. In one of the most common examples, hemoglobin S, the α chains are normal but the β chains have a single substitution of a valine residue for one glutamic acid, leading to sickle cell anemia (Table 31–4). When an abnormal gene inherited from one parent dictates formation of an abnormal hemoglobin (ie, when the individual is heterozygous), half the circulating hemoglobin is abnormal and half is normal. When identical abnormal genes are inherited from both parents, the individual is homozygous and all the hemoglobin is abnormal. It is theoretically possible to inherit two different abnormal hemoglobins, one from the father and one from the mother. Studies of the inheritance and geographic distribution of abnormal hemoglobins have made it possible in some cases to decide where the mutant gene originated and approximately how long ago the mutation occurred. In general, harmful mutations tend to die out, but mutant genes that confer traits with survival value persist and spread in the population. Many of the abnormal hemoglobins are harmless; however, some have abnormal O2 equilibriums, while others cause anemia. For example, hemoglobin S polymerizes at low O2 tensions, and this causes the red cells to become sickle-shaped, hemolyze, and form aggregates that block blood vessels. The sickle cell gene is an example of a gene that has persisted and spread in the population due to its beneficial effect when present in heterozygous form. It originated in Africa, and confers resistance to one type of malaria. In some parts of Africa, 40% of the population is heterozygous for hemoglobin S. There is a corresponding prevalence of 10% among African Americans in the United States.
THERAPEUTIC HIGHLIGHTSHemoglobin F decreases the polymerization of deoxygenated hemoglobin S, and hydroxyurea stimulates production of hemoglobin F in children and adults. Hydroxyurea has therefore proven to be a very valuable agent for the treatment of sickle cell disease. In patients with severe sickle cell disease, hematopoietic stem cell transplantation has also been shown to have some benefit, and prophylactic treatment with antibiotics has also been shown to be helpful. The clinically important thalassemias result in severe anemia, often requiring repeated blood transfusions. However, these run the risk of iron overload, and often must be accompanied by treatment with drugs that chelate iron. Hematopoietic stem cell transplantation is also being explored for treatment of the thalassemias.
| Positions on Polypeptide Chain of Hemoglobin | |||||||
|---|---|---|---|---|---|---|---|
| Hemoglobin | 1 2 3 | 6 7 | 26 | 63 | 67 | 121 | 146 |
| A (normal) | Val-His-Leu | Glu-Glu | Glu | His | Val | Glu | His |
| S (sickle cell) | Val | ||||||
| C | Lys | ||||||
| GSan Jose | Gly | ||||||
| E | Lys | ||||||
| MSaskatoon | Tyr | ||||||
| MMilwaukee | Glu | ||||||
| OArabia | Lys | ||||||
Incompatibilities in the ABO blood group system are summarized in Table 31–3. Persons with type AB blood are “universal recipients” because they have no circulating agglutinins and can be given blood of any type without developing a transfusion reaction due to ABO incompatibility. Type O individuals are “universal donors” because they lack A and B antigens, and type O blood can be given to anyone without producing a transfusion reaction due to ABO incompatibility. This does not mean, however, that blood should ever be transfused without being cross-matched except in the most extreme emergencies, since the possibility of reactions or sensitization due to incompatibilities in systems other than ABO systems always exists. In cross-matching, donor red cells are mixed with recipient plasma on a slide and checked for agglutination. It is advisable to check the action of the donor’s plasma on the recipient cells in addition, even though, as noted above, this is rarely a source of trouble.
A procedure that has recently become popular is to withdraw the patient’s own blood in advance of elective surgery and then infuse this blood back (autologous transfusion) if a transfusion is needed during the surgery. With iron treatment, 1000–1500 mL can be withdrawn over a 3-weeks period. The popularity of banking one’s own blood is primarily due to fear of transmission of infectious diseases by heterologous transfusions, but of course another advantage is elimination of the risk of transfusion reactions.
The A and B antigens are inherited as mendelian allelomorphs, A and B being dominants. For example, an individual with type B blood may have inherited a B antigen from each parent or a B antigen from one parent and an O from the other; thus, an individual whose phenotype is B may have the genotype BB (homozygous) or BO (heterozygous).
When the blood types of the parents are known, the possible genotypes of their children can be stated. When both parents are type B, they could have children with genotype BB (B antigen from both parents), BO (B antigen from one parent, O from the other heterozygous parent), or OO (O antigen from both parents, both being heterozygous). When the blood types of a mother and her child are known, typing can prove that a man cannot be the father, although it cannot prove that he is the father. The predictive value is increased if the blood typing of the parties concerned includes identification of antigens other than the ABO agglutinogens. With the use of DNA fingerprinting (see Chapter 1), the exclusion rate for paternity rises to close to 100%.
In addition to the ABO system of antigens in human red cells, there are systems such as the Rh, MNSs, Lutheran, Kell, Kidd, and many others. There are over 500 billion possible known blood group phenotypes, and because undiscovered antigens undoubtedly exist, it has been calculated that the number of phenotypes is actually in the trillions.
The number of blood groups in animals is as large as it is in humans. An interesting question is why this degree of polymorphism developed and has persisted through evolution. Certain diseases are more common in individuals with one blood type or another, but the differences are not great. The significance of a recognition code of this complexity is therefore unknown.
Aside from the antigens of the ABO system, those of the Rh system are of the greatest clinical importance. The Rh factor, named for the rhesus monkey because it was first studied using the blood of this animal, is a system composed primarily of the C, D, and E antigens, although it actually contains many more. Unlike the ABO antigens, the system has not been detected in tissues other than red cells. D is by far the most antigenic component, and the term Rh-positive as it is generally used means that the individual has agglutinogen D. The D protein is not glycosylated, and its function is unknown. The Rh-negative individual has no D antigen and forms the anti-D agglutinin when injected with D-positive cells. The Rh typing serum used in routine blood typing is anti-D serum. Eighty-five percent of whites are D-positive and 15% are D-negative; over 99% of Asians are D-positive. Unlike the antibodies of the ABO system, anti-D antibodies do not develop without exposure of a D-negative individual to D-positive red cells by transfusion or entrance of fetal blood into the maternal circulation. However, D-negative individuals who have received a transfusion of D-positive blood (even years previously) can have appreciable anti-D titers and thus may develop transfusion reactions when transfused again with D-positive blood.
Another complication due to Rh incompatibility arises when an Rh-negative mother carries an Rh-positive fetus. Small amounts of fetal blood leak into the maternal circulation at the time of delivery, and some mothers develop significant titers of anti-Rh agglutinins during the postpartum period. During the next pregnancy, the mother’s agglutinins cross the placenta to the fetus. In addition, there are some cases of fetal-maternal hemorrhage during pregnancy, and sensitization can occur during pregnancy. In any case, when anti-Rh agglutinins cross the placenta to an Rh-positive fetus, they can cause hemolysis and various forms of hemolytic disease of the newborn (erythroblastosis fetalis). If hemolysis in the fetus is severe, the infant may die in utero or may develop anemia, severe jaundice, and edema (hydrops fetalis). Kernicterus, a neurologic syndrome in which unconjugated bilirubin is deposited in the basal ganglia, may also develop, especially if birth is complicated by a period of hypoxia. Bilirubin rarely penetrates the brain in adults, but it does in infants with erythroblastosis, possibly in part because the blood–brain barrier is more permeable in infancy. However, the main reasons that the concentration of unconjugated bilirubin is very high in this condition are that production is increased and the bilirubin-conjugating system is not yet mature.
About 50% of Rh-negative individuals are sensitized (develop an anti-Rh titer) by transfusion of Rh-positive blood. Because sensitization of Rh-negative mothers by carrying an Rh-positive fetus generally occurs at birth, the first child is usually normal. However, hemolytic disease occurs in about 17% of the Rh-positive fetuses born to Rh-negative mothers who have previously been pregnant one or more times with Rh-positive fetuses. Fortunately, it is usually possible to prevent sensitization from occurring the first time by administering a single dose of anti-Rh antibodies in the form of Rh immune globulin during the postpartum period. Such passive immunization does not harm the mother and has been demonstrated to prevent active antibody formation by the mother. In obstetric clinics, the institution of such treatment on a routine basis to unsensitized Rh-negative women who have delivered an Rh-positive baby has reduced the overall incidence of hemolytic disease by more than 90%. In addition, fetal Rh typing with material obtained by amniocentesis or chorionic villus sampling is now possible, and treatment with a small dose of Rh immune serum will prevent sensitization during pregnancy.
PLASMA
The fluid portion of the blood, the plasma, is a remarkable solution containing an immense number of ions, inorganic molecules, and organic molecules that are in transit to various parts of the body or aid in the transport of other substances. Normal plasma volume is about 5% of body weight, or roughly 3500 mL in a 70-kg man. Plasma clots on standing, remaining fluid only if an anticoagulant is added. If whole blood is allowed to clot and the clot is removed, the remaining fluid is called serum. Serum has essentially the same composition as plasma, except that its fibrinogen and clotting factors II, V, and VIII (Table 31–5) have been removed and it has a higher serotonin content because of the breakdown of platelets during clotting.
| Factora | Names |
|---|---|
| I | Fibrinogen |
| II | Prothrombin |
| III | Thromboplastin |
| IV | Calcium |
| V | Proaccelerin, labile factor, accelerator globulin |
| VII | Proconvertin, SPCA, stable factor |
| VIII | Antihemophilic factor (AHF), antihemophilic factor A, antihemophilic globulin (AHG) |
| IX | Plasma thromboplastic component (PTC), Christmas factor, antihemophilic factor B |
| X | Stuart-Prower factor |
| XI | Plasma thromboplastin antecedent (PTA), antihemophilic factor C |
| XII | Hageman factor, glass factor |
| XIII | Fibrin-stabilizing factor, Laki-Lorand factor |
| HMW-K | High-molecular-weight kininogen, Fitzgerald factor |
| Pre-Ka | Prekallikrein, Fletcher factor |
| Ka | Kallikrein |
| PL | Platelet phospholipid |
The plasma proteins consist of albumin, globulin, and fibrinogen fractions. Most capillary walls are relatively impermeable to the proteins in plasma, and the proteins therefore exert an osmotic force of about 25 mm Hg across the capillary wall (oncotic pressure; see Chapter 1) that pulls water into the blood. The plasma proteins are also responsible for 15% of the buffering capacity of proteins in the blood (including hemoglobin; see Chapter 39) because of the weak ionization of their substituent COOH and NH2 groups. At the normal plasma pH of 7.40, the proteins are mostly in the anionic form (see Chapter 1). Plasma proteins may have specific functions (eg, antibodies and the proteins concerned with blood clotting), whereas others function as nonspecific carriers for various hormones, other solutes, and drugs.
Circulating antibodies are manufactured by lymphocytes. Most of the other plasma proteins are synthesized in the liver. These proteins and their principal functions are listed in Table 31–6.
| Name | Principal Function | Binding Characteristics | Serum or Plasma Concentration |
|---|---|---|---|
| Albumin | Binding and carrier protein; osmotic regulator | Hormones, amino acids, steroids, vitamins, fatty acids | 4500–5000 mg/dL |
| Orosomucoid | Uncertain; may have a role in inflammation | Trace; rises in inflammation | |
| α1-Antiprotease | Trypsin and general protease inhibitor | Proteases in serum and tissue secretions | 1.3–1.4 mg/dL |
| α-Fetoprotein | Osmotic regulation; binding and carrier proteina | Hormones, amino acids | Found normally in fetal blood |
| α2-Macroglobulin | Inhibitor of serum endoproteases | Proteases | 150–420 mg/dL |
| Antithrombin-III | Protease inhibitor of intrinsic coagulation system | 1:1 binding to proteases | 17–30 mg/dL |
| Ceruloplasmin | Transport of copper | Six atoms copper/molecule | 15–60 mg/dL |
| C-reactive protein | Uncertain; has role in tissue inflammation | Complement C1q | <1 mg/dL; rises in inflammation |
| Fibrinogen | Precursor to fibrin in hemostasis | 200–450 mg/dL | |
| Haptoglobin | Binding, transport of cell-free hemoglobin | Hemoglobin 1:1 binding | 40–180 mg/dL |
| Hemopexin | Binds to porphyrins, particularly heme for heme recycling | 1:1 with heme | 50–100 mg/dL |
| Transferrin | Transport of iron | Two atoms iron/molecule | 3.0–6.5 mg/dL |
| Apolipoprotein B | Assembly of lipoprotein particles | Lipid carrier | |
| Angiotensinogen | Precursor to pressor peptide angiotensin II | ||
| Proteins, coagulation factors II, VII, IX, X | Blood clotting | 20 mg/dL | |
| Antithrombin C, protein C | Inhibition of blood clotting | ||
| Insulinlike growth factor I | Mediator of anabolic effects of growth hormone | IGF-I receptor | |
| Steroid hormone-binding globulin | Carrier protein for steroids in bloodstream | Steroid hormones | 3.3 mg/dL |
| Thyroxine-binding globulin | Carrier protein for thyroid hormone in bloodstream | Thyroid hormones | 1.5 mg/dL |
| Transthyretin (thyroid-binding prealbumin) | Carrier protein for thyroid hormone in bloodstream | Thyroid hormones | 25 mg/dL |
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


