Learning Objectives
Learn the basic molecular events in clot formation and fibrinolysis.
Understand the basic classification of disorders in hemostasis.
Identify the appropriate laboratory tests for evaluation of the bleeding patient and the thrombotic patient.
Learn the prominent clinical and laboratory features of the individual disorders of hemostasis.
The coagulopathies are grouped into disorders of bleeding and thrombosis. The hemorrhagic diseases are further subdivided into the 2 major categories of coagulation factor disorders and platelet disorders. To understand the diseases with abnormal coagulation that follow, a brief introduction to normal hemostasis precedes the discussions of the diseases.
Introduction to Hemostasis
Normal hemostasis is the controlled activation of coagulation factors and platelets leading to clot formation, with subsequent clot lysis, in a process that stops hemorrhage without excess clotting (thrombosis). Effective hemostasis is a rapid and localized response to an interruption in vascular integrity (vessel wall injury), such that clots are formed only when and where they are needed.
Clot formation involves platelet activation and the subsequent generation of fibrin via the coagulation cascade. The 2 processes are discussed separately in the sections that follow.
Platelet plug formation is initiated in vivo by exposure of platelets to vascular subendothelium when a vessel is injured. The platelets adhere to the subendothelium, spread out along the surface, and release substances that promote the aggregation of other platelets at that site. The platelets also accelerate fibrin clot formation by providing a reactive surface for several steps in the coagulation cascade.
Clot formation involves platelet activation and the subsequent generation of fibrin via the coagulation cascade.
Adhesion of platelets to the subendothelial surface is facilitated by a plasma protein, von Willebrand factor (vWF), especially in vessels with high shear forces (eg, the fast blood flow in arteries has a higher shear force than slow blood flow in veins). vWF binds to a specific receptor on the platelet surface. Deficiency of vWF results in poor adherence of platelets to subendothelium. The severity of bleeding in von Willebrand disease (vWD) varies widely among patients. Another related platelet adhesion defect occurs in patients whose platelets lack the receptor for vWF. This bleeding disorder, known as Bernard–Soulier disease, results from an inability of platelets to bind vWF.
Platelet activation occurs from interaction of platelet agonists, most of which are soluble, with specific receptors on the platelet membrane. Physiologically important agonists include adenosine diphosphate (ADP), thrombin, epinephrine, collagen, and thromboxane A2, which is derived from arachidonic acid. A sequence of membrane and cytoplasmic events is initiated by the agonist–receptor interaction, involving an increase in cytoplasmic calcium ion concentration and a platelet shape change from a disc to a spiny sphere. The change in cytoplasmic calcium concentration leads to contractile events in the platelet, causing alpha and delta granules (also known as dense bodies) to fuse with the platelet plasma membrane and release their granule contents into the extracellular space. Successful granule release requires the formation of thromboxane A2 from endogenous arachidonate via the enzyme cyclooxygenase. This enzyme is inhibited irreversibly by aspirin and inhibited reversibly by a number of other anti-inflammatory agents. Treatment with aspirin can cause a platelet secretion defect (reduced release of granule contents) that is often clinically significant in patients with underlying coagulopathies. Alpha granules contain vWF, fibrinogen, Factor V, 2 platelet-specific proteins—platelet factor 4 (PF4) and beta–thromboglobulin—as well as a number of other proteins. Delta granules contain serotonin, adenosine triphosphate (ATP), ADP, pyrophosphate, polyphosphate, and calcium. The release of some of these substances, in particular ADP, activates unstimulated platelets nearby. Absence or deficiency of alpha or delta granules occurs as a feature of several congenital and acquired platelet function disorders, collectively known as storage pool disorders. Individuals whose platelets possess appropriate numbers of intact granules that cannot be released on appropriate stimulation have a platelet release defect, and on that basis also may have a bleeding tendency. Aspirin is a common cause of a platelet release defect, because it impairs thromboxane production.
Release of platelet granule contents is followed by platelet aggregation, that is, the binding of platelets to one another to form the platelet plug. Normal aggregation requires fibrinogen binding to platelets via the fibrinogen receptor, which is the glycoprotein IIb/IIIa complex (GP IIb/IIIa), on the platelet surface. Congenital absence of GP IIb/IIIa results in a bleeding diathesis known as Glanzmann thrombasthenia (GT).
The platelet surface serves as a site for certain coagulation pathway enzyme reactions (see below). For example, the platelet membrane can bind the Factor Xa/Factor Va complex that activates prothrombin to thrombin. Thus, platelet activation and fibrin formation via the coagulation cascade are interactive biological processes.
The coagulation factor pathway is an enzymatic cascade with sequential conversion of proenzymes (zymogens) to fully activated enzymes, which then convert other zymogens to their activated forms (Figure 11–1). The final steps directly preceding fibrin formation can be activated through both the intrinsic and extrinsic pathways; hence, this part of the pathway is called the common pathway. Numerous positive and negative feedback mechanisms exist in the coagulation pathways so that the cascades do not proceed in an uncontrolled fashion. The pathways are now known to be extremely complex, with multiple interactions between factors in the intrinsic, extrinsic, and common pathways. The following description is a version of coagulation factor interactions that highlights the fundamental reactions.
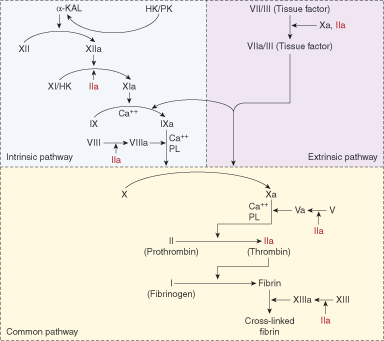
The coagulation cascade is activated by the appearance of tissue factor (also historically known as Factor III), which is not normally exposed. Tissue factor is presented when a blood vessel is injured. It binds with Factor VII and small amounts of circulating active Factor VII (Factor VIIa), resulting in a complex of Factor VII or VIIa and tissue factor. The Factor VII in the complex can be autoconverted to Factor VIIa, resulting in a greater number of Factor VIIa–tissue factor complexes. This complex activates Factor IX to Factor IXa in the intrinsic pathway, with some activation of Factor X to Factor Xa in the common pathway. Factor IXa, with support from Factor VIII as a cofactor (or Factor VIIIa, which is a much more effective cofactor), activates Factor X to Factor Xa. Factor Xa with the assistance of Factor V or, more effectively Factor Va, converts prothrombin (Factor II) to thrombin (Factor IIa). At this point, the coagulation cascade is markedly amplified because thrombin activates Factor VIII to Factor VIIIa, a more effective cofactor, and activates Factor V to its more effective Factor Va form. In addition, thrombin activates Factor XI to Factor XIa, which, like Factor VIIa and tissue factor, activates Factor IX to Factor IXa. Thrombin catalyzes the conversion of fibrinogen to fibrin, which is then cross-linked by Factor XIII to create a stabilized form of fibrin (clot). Factor XIII is also activated to Factor XIIIa by thrombin.
Factor XII, prekallikrein (PK), and high-molecular-weight kininogen (HMWK, shown as HK in Figure 11–1) are not required for the generation of fibrin in vivo because even when they are completely absent, there is no increased risk for bleeding. Nevertheless, the intrinsic pathway is activated when Factor XII contacts collagen (exposed by vessel injury) or polyphosphate (released from activated platelets).
This cascade, as it is currently understood and described above, explains 2 long-standing clinical observations. First, it explains the significant bleeding tendency associated with Factors VIII and IX deficiencies because these factors are important in the early stages of cascade amplification. Factor VIIa and tissue factor activate Factor IX to Factor IXa, and Factor IXa requires Factor VIII to convert Factor X to Factor Xa. Second, this scheme provides an explanation for the clinical observation that deficiencies of Factor XII, PK, and HMWK are not associated with an increased risk for bleeding because Factor VIIa/TF activates Factor IX, and thrombin activates Factor XI, bypassing the need for Factor XII.
Two of the coagulation cascade reactions occur on the platelet surface. The first of these is the activation of Factor X to Factor Xa, which is produced by platelet-bound Factor IXa and platelet-bound Factor VIIIa. In the second, platelet-bound Factor Xa and platelet-bound Factor Va convert prothrombin (Factor II) to thrombin (Factor IIa) in a subsequent step in the coagulation sequence. As noted below, single factor deficiency states, most of which are congenital, exist for all of the factors, but multiple factor deficiencies, which are usually not congenital, are much more commonly encountered. Inhibitors, as antibodies directed against a specific coagulation factor, can arise to any of the individual factors to create deficiency states. With some exceptions, most factor inhibitors are rare.
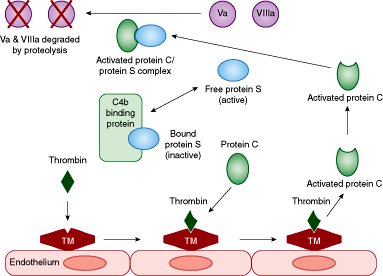
There are 2 major inhibitory pathways that determine the rate at which the cascade is amplified. One of these is the protein C–protein S anticoagulant pathway. An additional mechanism for control of the coagulation cascade involves the inhibitory action of antithrombin.
There are 2 major inhibitory pathways that determine the rate at which the cascade is amplified. One of these is the protein C–protein S anticoagulant pathway (Figure 11–2). As shown in the figure, excess thrombin, which is generated through the activation of the coagulation cascade, provides the signal to shut off the coagulation cascade by binding to a protein on the endothelial surface known as thrombomodulin. The thrombin/thrombomodulin complex converts protein C into its activated form. The activated protein C then binds free protein S as a cofactor. Protein S may be bound to C4b-binding protein and to a limited number of other proteins, in which case protein S becomes inactive. Once free (unbound) protein S binds to activated protein C, the activated protein C/protein S complex then proteolytically degrades Factors Va and VIIIa, reducing the flux through the coagulation cascade by removing these 2 activated cofactors. A mutation in the Factor V molecule, known as the Factor V Leiden mutation, makes Factor V resistant to proteolytic degradation by the activated protein C/protein S complex. This condition is known as activated protein C resistance. This permits the prothrombotic action of Factor Va to persist and contribute to a hypercoagulable state.
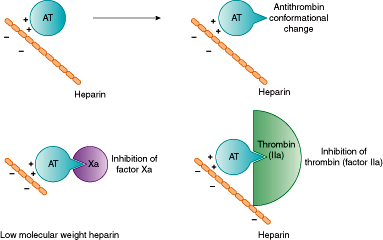
An additional mechanism for control of the coagulation cascade involves the inhibitory action of antithrombin (formerly known as antithrombin III) (Figure 11–3). Antithrombin has a limited anticoagulant effect on its own, but in the presence of heparin or selected other negatively charged heparin-like molecules, antithrombin adopts a new conformation that increases its inhibitory activity 1000-fold, permitting it to inhibit most of the activated coagulation factors in complexes where both antithrombin and heparin bind to the activated coagulation factor. Inhibition of Factor Xa, however, does not require the direct binding of the activated coagulation factor by heparin. Factor Xa can be neutralized when it is bound only to antithrombin, after antithrombin has been activated by heparin or related molecule. The antithrombotic action of short chains of heparin (low-molecular-weight heparin) is directed primarily against Factor Xa because short heparin chains inhibit predominantly only Factor Xa (longer chains are required for Factor IIa inhibition). Fondaparinux, a synthetic heparin-related pentasaccharide, inhibits Factor Xa exclusively.
It should be noted that all of the coagulation factors in the coagulation cascade are synthesized in the liver, and that the activity of Factors II, VII, IX, and X is vitamin K-dependent. Tissue factor is constitutively expressed on some cell types, and in other cell types, such as endothelial cells, it is not normally expressed. There is little (if any) tissue factor in the circulating plasma under normal conditions. It is not exclusively synthesized in the liver. Tissue factor in the subendothelium is exposed to blood when blood vessels are injured, triggering the extrinsic pathway of the coagulation cascade. The half-lives of the coagulation factors in the blood vary markedly, with Factor VII showing the shortest half-life of approximately 5 hours and Factor XIII having the longest half-life of more than 120 hours. There is also a wide range of plasma concentrations for the coagulation factors. Factor VII is in the lowest concentration of the circulating coagulation factors at 100 to 500 ng/mL. The highest concentration is found for fibrinogen at 200 to 400 mg/dL.
Fibrinolysis is the controlled dissolution of the formed clot. It occurs when the injured vessel begins to heal, and is initiated to a limited extent when clot formation begins. In this way, fibrinolysis serves as a regulatory mechanism to limit excess clot formation.
Fibrinolysis is the controlled dissolution of the formed clot. It occurs when the injured vessel begins to heal, and is initiated to a limited extent when clot formation begins. In this way, fibrinolysis serves as a regulatory mechanism to limit excess clot formation.
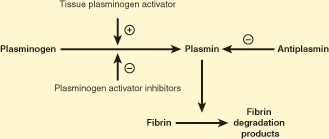
The principal enzyme involved in fibrinolysis is plasmin, which exists in a zymogen form known as plasminogen (Figure 11–4). Plasminogen is converted into plasmin by tissue plasminogen activator (tPA). Plasmin degrades the fibrin clot. A recombinant form of tPA is used as a pharmacologic agent to produce thrombolysis (breakdown of a thrombus), in patients with myocardial infarction or stroke, and clots elsewhere in the body. Clinically effective derivatives of tPA from genetic manipulation are also used for thrombolysis. tPA is released by endothelial cells, and its secretion into the plasma is increased by thrombin. Plasminogen activator inhibitors are secreted by platelets and endothelial cells, particularly when they are activated. Plasminogen activator inhibitors stabilize a newly formed clot by blocking the action of tPA, thereby preventing premature dissolution of the clot. Plasmin degrades fibrin polymers, and, to a limited degree, fibrinogen as well, by specific and sequential proteolytic cleavages, generating fibrin degradation products (FDP). These FDP (fragments X, Y, D, D-dimer, and E) may be detected in the plasma of patients experiencing fibrinolysis. Deficiency of plasminogen may predispose to thrombosis, and deficiency of plasminogen activator inhibitor may increase the risk for bleeding. Antiplasmin inhibits plasmin, but only when it is circulating and not when it is clot-bound. This prevents the circulation of a proteolytically active form of plasmin, while permitting clot lysis to proceed by plasmin within the clot. Deficiency of antiplasmin may result in a hemorrhagic tendency.
Figure 11–4
The fibrinolytic pathway. A plus sign indicates that tissue plasminogen activator converts plasminogen into plasmin. A minus sign indicates inhibitory action. (Redrawn with permission from Kabakibi, A et al. The hypercoagulable state.Turnaround Times [a newsletter for physicians at the Massachusetts General Hospital, Boston]. 1994;3:1:1.)
Bleeding Disorders
Figure 11–5 provides a classification of coagulation disorders. The major division in the classification is between disorders associated with bleeding and disorders associated with thrombosis. There are 2 major subdivisions of bleeding disorders—those associated with coagulation factor and fibrinolytic pathway factor deficiencies, and those associated with an abnormal platelet count or impaired platelet function. Isolated factor deficiencies are usually congenital, although occasionally an isolated acquired factor deficiency develops. An example of an acquired isolated coagulation factor deficiency is the Factor X deficiency associated with amyloidosis. The deficiency of antiplasmin is listed in this section because its absence permits increased plasmin activity and overactive clot dissolution, resulting in a bleeding tendency. Another major category of coagulation factor abnormalities is multiple coagulation factor deficiencies. There are several commonly encountered situations associated with multiple factor deficiencies. These include vitamin K deficiency or warfarin intake (which results in a reduced amount of functional Factors II, VII, IX, and X as well as protein C and protein S); disseminated intravascular coagulation (DIC), which results in the consumption of multiple coagulation factors; and liver disease that results in decreased synthesis of coagulation factors. Several activated coagulation factors are inhibited by heparin. Heparin administration results in inactivation of most of the activated coagulation factors.
Quantitative platelet disorders include thrombocytopenia and thrombocytosis. Qualitative platelet disorders are characterized by abnormal platelet function in the presence of a normal platelet count.
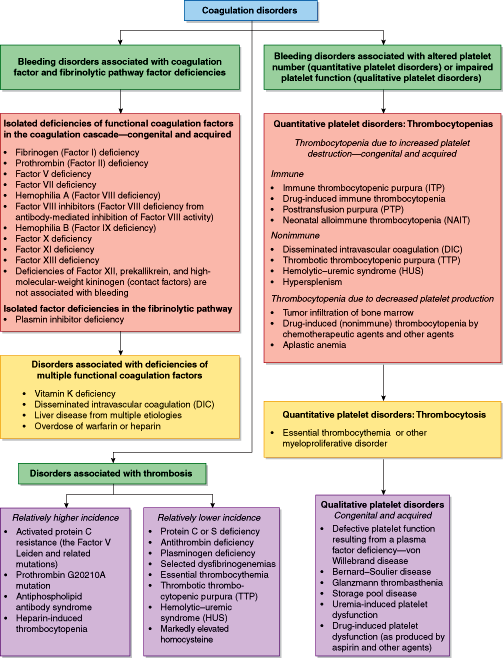
The group of disorders associated with platelets is divided first into quantitative platelet disorders and qualitative platelet disorders. Quantitative platelet disorders include thrombocytopenia and thrombocytosis. Thrombocytopenia can be produced as a result of increased platelet destruction, from a variety of immune or nonimmune causes, or decreased platelet production. Common causes of decreased platelet production include tumor infiltration of bone marrow from metastases or a hematologic malignancy, and drug-induced thrombocytopenia as occurs with chemotherapy. Thrombocytopenia can also occur as a result of increased sequestration of platelets in the spleen, usually in patients with splenomegaly. Thrombocytosis is much less common than thrombocytopenia. Thrombocytosis can be divided into reactive thrombocytosis, in which there is a transiently increased number of platelets from a stimulus to increase platelet production, or neoplastic thrombocytosis, as seen in myeloproliferative disease and, less commonly, myelodysplastic disorders.
Qualitative platelet disorders are characterized by abnormal platelet function in the presence of a normal platelet count. vWD is a disorder in which there is defective platelet function, but from a defect originating outside the platelet, since vWF is generated primarily in endothelial cells. vWF coats the surface of the activated platelet to allow it to adhere to the cut vessel surface and initiate platelet plug formation.
Other causes of defective platelet function result from abnormalities within the platelet. These disorders may be congenital or acquired. The congenital ones are extremely rare, and the acquired ones are very frequently encountered. Congenital platelet abnormalities associated with defective function include GT, Bernard–Soulier disease, and storage pool disease (SPD). The much more common acquired qualitative platelet disorders include drug-induced platelet dysfunction, such as produced by aspirin and clopidogrel (Plavix), and uremia-induced platelet dysfunction, which occurs in patients with impaired renal function.
Fibrinogen is produced in the liver by hepatocytes. Abnormalities of fibrinogen production may be congenital or acquired and, in general, involve either decreased production of a normal molecule (afibrinogenemia and hypofibrinogenemia) or production of an abnormal molecule (dysfibrinogenemia) (see Table 11–1).
| Results/Comments | ||||
|---|---|---|---|---|
| Quantitative Deficiencies | Dysfibrinogenemia (Qualitative Deficiencies) | |||
| Laboratory Test | Afibrinogenemia | Hypofibrinogenemia | Homozygous | Heterozygous |
| PT | Markedly prolonged | Normal to slightly prolonged | Markedly prolonged | Slightly prolonged to normal |
| PTT | Markedly prolonged | Normal to slightly prolonged | Markedly prolonged | Slightly prolonged to normal |
| Functional fibrinogen | Low | Slightly low to normal | Low | Slightly low to normal |
| Immunologic fibrinogen | Low | Slightly low to normal | Normal | Normal |
| Thrombin time | Prolonged | Normal to prolonged | Prolonged | Prolonged |
| Reptilase time | Prolonged | Normal to prolonged | Prolonged | Prolonged |
In congenital afibrinogenemia and hypofibrinogenemia, there is a reduced (hypofibrinogenemia) or absent (afibrinogenemia) production of a normal fibrinogen molecule. In general, the homozygous deficiency results in afibrinogenemia, and the heterozygous state results in hypofibrinogenemia. Both disorders are rare. Homozygotes suffer a mild-to-moderate spontaneous bleeding tendency. Manifestations include umbilical stump hemorrhage and bleeding from mucous membranes, among many other possible signs and symptoms related to blood loss. Severe bleeding may occur with trauma or surgery. Hypofibrinogenemic patients are usually asymptomatic, but may bleed significantly with surgery or trauma.
Congenital dysfibrinogenemia is a result of inheritance of a gene for an abnormal fibrinogen molecule, which is produced in normal or near-normal quantities. All the fibrinogen produced by a homozygote for dysfibrinogenemia is abnormal, and approximately half of the fibrinogen in a heterozygote is abnormal. Hundreds of abnormal fibrinogens have been described. The true incidence of dysfibrinogenemia is not known because many forms of the disorder are asymptomatic. Homozygotes may have a mild bleeding tendency, perhaps because the fibrinogen molecule is cleaved too slowly to form fibrin monomers or because abnormal fibrin monomers polymerize too slowly. The bleeding tendency is characterized by easy or spontaneous bruising, menorrhagia, and prolonged or severe bleeding with surgery or trauma. Heterozygotes are usually asymptomatic, but may show excessive bleeding with surgery or trauma. Several types of dysfibrinogenemia (approximately 10%-15% of cases) are associated with an increased risk of thrombosis rather than bleeding. A few types of congenital dysfibrinogenemia are associated with both bleeding and thrombosis.
Acquired hypofibrinogenemia is observed predominantly in patients with advanced liver disease, in patients with a consumptive coagulation disorder such as DIC, and in those treated with thrombolytic therapy.
Acquired hypofibrinogenemia is observed predominantly in patients with advanced liver disease, in patients with a consumptive coagulation disorder such as DIC, and in those treated with thrombolytic therapy.
Acquired dysfibrinogenemia represents the acquired production of an abnormal fibrinogen molecule in normal or near-normal quantities, most often in patients with acute or chronic liver disease, especially those with primary or metastatic hepatic malignancies. The patient may or may not be symptomatic, depending on 1) whether there is simultaneous production of normal fibrinogen in amounts sufficient to allow normal hemostasis and 2) whether the abnormal fibrinogen can polymerize like a normal fibrinogen molecule (see the section “Hemostatic Abnormalities in Liver Disease”).
See Table 11–1 for the laboratory evaluation of the patient with a fibrinogen deficiency.
Prothrombin (Factor II) is the precursor to thrombin (Factor IIa), which converts fibrinogen into fibrin in the common pathway of the coagulation cascade. Deficiency of prothrombin, either inherited or acquired, may result in a hemorrhagic diathesis. Inherited abnormalities of prothrombin are rare. As with fibrinogen, abnormalities occur in 2 major forms. The first is reduced or absent production of a normal prothrombin molecule. The second is production of normal amounts of an abnormal prothrombin molecule with decreased activity (dysfunctional form or dysprothrombinemia). Heterozygotes usually have approximately 50% of normal activity and may be asymptomatic or have a bleeding tendency. In 1 study, 83% of a small cohort of heterozygotes had bleeding, with Factor II levels ranging from 21% to 35%. Homozygotes usually have 1% to 25% of normal activity and have a mild-to-severe hemorrhagic tendency. Acquired hypoprothrombinemia occurs most often along with deficiencies of Factors VII, IX, and X in vitamin K deficiency and with warfarin (Coumadin) therapy; with deficiencies of multiple coagulation factors in liver disease or DIC; as an isolated coagulation factor deficiency in some patients with lupus anticoagulant (LA); and in patients exposed to topical bovine thrombin who develop antibodies to prothrombin (and not uncommonly to Factor V also). Bleeding manifestations depend on the level of prothrombin activity; usually no bleeding occurs with a prothrombin level >50% of normal.
Abnormalities occur in 2 major forms. The first is reduced or absent production of a normal prothrombin molecule. The second is production of normal amounts of an abnormal prothrombin molecule with decreased activity (dysfunctional form or dysprothrombinemia).
See Table 11–2 for the laboratory evaluation of the patient with a prothrombin (Factor II) deficiency.
| Deficient Factor(s) | PT Prolonged If Deficient? | PTT Prolonged If Deficient? | Other Tests Useful in Diagnosis/Comments |
|---|---|---|---|
| Fibrinogen—see Table 11–1 | |||
| Prothrombin (Factor II) | Yes | Yes (usually less prominent than the PT prolongation) |
|
| Factor V | Yes | Yes (usually less prominent than the PT prolongation) |
|
| Factor VII | Yes | No | Factor VII assay; selected other factor assays to determine if Factor VII is low along with other factors, especially II, V, IX, and X |
| Factor VIII | No | Yes |
|
| Factor IX | No | Yes | Factors VIII and IX assays as these 2 deficiency states are clinically indistinguishable; selected other factor assays to determine if Factor IX is low along with other factors, especially II, VII, and X |
| Factor X | Yes | Yes (usually less prominent than the PT prolongation) | Factor X assay; selected other factor assays to determine if Factor X is low along with other factors, especially II, VII, and IX |
| Factor XI | No | Yes | Factor XI assay |
| Factor XII | No | Yes | Factor XII assay |
| Prekallikrein | No | Yes | Prekallikrein assay |
| High-molecular-weight kininogen | No | Yes | High-molecular-weight kininogen assay |
| Factor XIII | No | No | The screening test for Factor XIII deficiency assesses the solubility of the patient’s clot in urea—clots from patients with Factor XIII levels <2% dissolve; a quantitative assay not involving clot solubility is also available and can detect mild deficiencies of Factor XIII |
Factor V is a high-molecular-weight protein (approximately 300,000 Da) that acts as an accelerating cofactor for the enzymatic conversion of prothrombin to thrombin by Factor Xa. When Factor V is cleaved to Factor Va by thrombin, its cofactor activity is significantly increased. Factors Va and VIIIa are degraded by activated protein C. An isolated deficiency of Factor V is a rare cause of bleeding.
Apparent heterozygous and homozygous deficient states have been observed. Heterozygotes usually have levels of approximately 50% of normal and can experience bleeding or may be asymptomatic. In a cohort of 19 heterozygous patients, 50% had bleeding, with Factor V levels ranging from 21% to 55%. Homozygotes have variable levels below 50%; they are most likely to be symptomatic if the level is 10% or less.
As with the other coagulation factors, 2 major forms of the inherited deficiency are described: reduced or absent production of a normal Factor V molecule (absence form) and production of an abnormal molecule with reduced activity in normal amounts (dysfunctional form). A rare combined deficiency of Factors V and VIII is due to a genetic defect in intracellular transport of Factors V and VIII. Acquired deficiencies of Factor V occur with liver dysfunction or DIC.
See Table 11–2 for the laboratory evaluation of the patient with a Factor V deficiency.
Factor VII is a vitamin K-dependent coagulation factor precursor that, when activated by thrombin, Factor Xa, or Factor IXa, is converted to Factor VIIa. This activated factor then converts phospholipid-bound Factor X into Factor Xa in the presence of calcium and tissue factor. It also converts Factor IX to Factor IXa. Factor VII deficiency may occur as an inherited or acquired disorder.
The inherited deficiency state, which is rare, may be present as reduced or absent production of a normal molecule (absence form) or production of an abnormal molecule with decreased activity in normal amounts (dysfunctional form). An inherited isolated deficiency of Factor VII in heterozygotes is usually associated with a Factor VII activity level of approximately 50%. In a cohort of 88 heterozygous patients, 36% had a bleeding tendency, with Factor VII levels ranging from 21% to 69%. In homozygotes, there are variable Factor VII activity levels below 50%. The bleeding risk is difficult to predict in these patients because the factor activity level correlates poorly with the patient’s tendency to hemorrhage, but in general values <10% can be associated with major spontaneous bleeding. A large proportion of patients with less than 2% Factor VII do not bleed. Acquired Factor VII deficiency occurs in vitamin K deficiency and with warfarin therapy along with deficiencies of Factors II, IX, and X; and in DIC or liver disease along with multiple other coagulation factor deficiencies.
The bleeding risk is difficult to predict because the factor VII activity level correlates poorly with the patient’s tendency to hemorrhage. A large proportion of patients with less than 2% Factor VII do not bleed.
Intracranial hemorrhage has been reported in Factor VII-deficient patients, most often occurring in infants <1 year of age. Elevated Factor VII levels have been associated with an increased risk of cardiovascular disease.
See Table 11–2 for the laboratory evaluation of the patient with a Factor VII deficiency.
Hemophilia A is a bleeding disorder resulting from a deficiency of Factor VIII procoagulant activity. Factor VIII circulates in the plasma bound to vWF. Approximately 90% of patients with hemophilia A synthesize low amounts of normal Factor VIII molecules, and 10% of patients with hemophilia A synthesize normal amounts of an abnormal (nonfunctional) Factor VIII. Hemophilia A is inherited as an X-linked trait, and 65% to 75% of patients have a positive family history. Disease prevalence in the United States is 1 in 10,000 males; the carrier state in females is rarely symptomatic. Hemophilia A and hemophilia B (Factor IX deficiency, see below) are clinically indistinguishable. The likelihood of hemorrhage depends on the amount of Factor VIII present; the majority of patients (approximately 50%-70% of hemophilia A patients) have severe disease. The severity of disease is categorized as follows:
In mild disease: the VIII level is 6% to 20% of normal, with rare spontaneous bleeding.
In moderate disease: the VIII level is 1% to 5% of normal, with occasional spontaneous bleeding.
In severe disease: the VIII level is <1% of normal, with frequent spontaneous bleeding.
Hemophilia A is a bleeding disorder resulting from a deficiency of Factor VIII procoagulant activity. Factor VIII inhibitors are antibodies, usually IgG, that bind to Factor VIII and inhibit its coagulant activity.
All hemophilia patients (A and B) may experience severe hemorrhage following trauma or surgery if there is no prior treatment to elevate the factor level. Bleeding that is characteristic of hemophilia (A and B) includes intra-articular (joint), intracranial, and intramuscular hemorrhage. The latter can produce a compartment compression syndrome. Easy bruising and prolonged bleeding after minor cuts and abrasions are also characteristic. The onset of hemorrhage is typically delayed following injury, and pathologic bleeding may occur hours after injury. Primary hemostasis (dependent on platelet plug formation) is intact, but secondary hemostasis (dependent on the fibrin clot generated by the coagulation cascade) is defective. Up to 15% of hemophilia A patients develop an inhibitor to Factor VIII at some time during the course of their disease (ie, an antibody against Factor VIII). The inhibitor develops only in those transfused with Factor VIII-containing products, and most often in patients with <1% Factor VIII. Factor VIII inhibitors may also spontaneously occur rarely in nonhemophiliacs (see the section “Factor VIII Inhibitors”).
See Table 11–2 for information regarding the laboratory evaluation for Factor VIII deficiency.
Factor VIII inhibitors are antibodies, usually IgG, that bind to Factor VIII and inhibit its coagulant activity.
Factor VIII inhibitors have been found in several clinical situations.
Inhibitors are diagnosed most commonly in patients with hemophilia A. Inhibitors occur in 10% to 15% of these patients and make the treatment of hemorrhage much more difficult. The vast majority of cases of Factor VIII inhibitors in hemophilia A patients occur in those with severe hemophilia A (<1% Factor VIII activity). Inhibitor formation is related to transfusion of exogenous Factor VIII, and usually develops before 100 treatment days if it appears. Two immune response patterns have been observed in hemophilia A patients. The first is a high response pattern. Inhibitors rise to a high titer in response to exposure to Factor VIII. The titer may not decline for months to years, even without further exposure to Factor VIII. Rapid anamnestic responses are often seen within 3 to 7 days of reexposure in these patients. In the second pattern, there is a low response. In addition, inhibitors usually remain at a low titer despite reexposure. They may occasionally disappear and reappear spontaneously. Little, if any, anamnestic response is likely found in a low responder.
Spontaneous inhibitors to Factor VIII can occur in the postpartum patient. Usually they
are recognized 2 to 5 months after the birth of the first child and disappear spontaneously after 12 to 18 months. However, the course is variable, and there are reports of death from hemorrhage in some patients. Antigenic differences between mother and fetus do not sufficiently explain the development of a Factor VIII inhibitor, and its cause remains unknown.
Inhibitors may occur in those with allergic and enhanced immunologic reactions, including patients with:
(a) Rheumatoid arthritis
(b) Systemic lupus erythematosus
(c) Reactions to drugs, such as penicillin, chloramphenicol, sulfonamides, and phenytoin
(d) Malignancy
(e) Asthma
(f) Crohn disease
(g) Ulcerative colitis
(h) Pemphigus
(i) Multiple myeloma
Inhibitors may appear in patients without any obvious underlying disorder. These are usually older individuals, and the inhibitor may remit in several months, persist for years, or disappear with immunosuppressive therapy.
In a hemophilia A patient, a poor response to treatment with Factor VIII concentrate may be the first indication that an inhibitor is present, or there may be an increased frequency of bleeding episodes. In nonhemophiliacs, development of a new hemorrhagic tendency is usually the presenting feature of a spontaneous Factor VIII inhibitor. The most favorable prognoses are for patients with low titer inhibitors, peripartum women, and patients without an underlying disorder.
Hemophilia B is an inherited hemorrhagic disorder resulting from a lack of procoagulant activity of Factor IX.
See Table 11–3 for information regarding the laboratory evaluation for a Factor VIII inhibitor.
| Laboratory Test | Results/Comments |
|---|---|
| PT | Normal |
| PTT | Prolonged; normalizes in a 1:1 PTT mixing study of patient plasma and normal plasma with a 0-min incubation (ie, PTT performed immediately after mixing), but becomes prolonged with a longer incubation of the mixed plasma (60 or 120 min) at 37°C; the PTT of the mixed plasma at 60-120 min incubation with a clinically significant inhibitor is typically at least 8 s longer than the PTT of the mixed plasma at 0-min incubation |
| Factor VIII activity | Decreased |
| Factor VIII inhibitor assay | Used for quantitation of inhibitor; inhibitor levels are expressed in Bethesda units (BU); 1 BU/mL is the amount of inhibitor that produces a 50% reduction in Factor VIII activity |
Hemophilia B is an inherited hemorrhagic disorder resulting from a lack of procoagulant activity of Factor IX. Factor IX is a vitamin K-dependent factor that, in its active form (Factor IXa), is a serine protease of the intrinsic pathway of the coagulation cascade. Approximately 70% to 90% of hemophilia B patients have a deficiency of a normal coagulant protein, and 10% to 30% produce an abnormal Factor IX that is nonfunctional. Inheritance is sex-linked, with affected males, and female carriers. Of hemophilia B patients, 60% to 70% have a positive family history for bleeding. The prevalence of hemophilia B is much less than that of hemophilia A. Approximately 1 in 50,000 males in the United States has hemophilia B versus 1 in 10,000 males with hemophilia A. The hemophilia B carrier state in the female is usually asymptomatic, as is the case with hemophilia A. Acquired Factor IX deficiency may occur along with deficiencies of Factors II, VII, and X in patients with vitamin K deficiency or those receiving warfarin therapy, and with deficiencies of other coagulation factors in patients with liver disease, DIC, or nephrotic syndrome.
As previously noted, hemophilia B is clinically indistinguishable from hemophilia A. The severity of hemorrhage depends on the amount of Factor IX activity present:
In mild disease: 6% to 20% of normal IX activity is present, with rare spontaneous bleeding.
In moderate disease: 1% to 5% of normal IX activity is present, with occasional spontaneous bleeding.
In severe disease: <1% of normal activity is present, with frequent spontaneous bleeding.
Profuse bleeding may occur in any hemophilia B patient with trauma or surgery if there is no prior treatment to elevate the factor level. Bleeding in hemophilia B resembles that found in hemophilia A and includes deep tissue bleeding, intra-articular bleeding (hemarthrosis), intracranial bleeding (which may be lethal), and intramuscular bleeding with potential compartment compression syndrome. Severe mucosal membrane bleeding can occur in hemophilia, particularly after dental surgery.
Inhibitors develop to Factor IX in 1% to 5% of hemophilia B cases. These antibodies often occur in high titer and frequently present a major bleeding problem despite treatment.
See Table 11–2 for information regarding the laboratory evaluation for patients with hemophilia B.
An inherited isolated deficiency of Factor X is a rare disorder. Homozygotes and heterozygotes have been identified. Homozygotes usually possess <2% of normal activity. Heterozygotes usually possess 40% to 70% of normal activity. In a cohort of 15 heterozygous patients, 33% had a bleeding tendency, with Factor X levels ranging from 23% to 47%. Patients with Factor X values <10% can have a high risk of spontaneous major bleeding, and those with >40% are usually asymptomatic. Inherited Factor X deficiency, like the other factor deficiency states, occurs in 2 major forms: reduced or absent synthesis of a normal molecule (absence form) and synthesis of an abnormal molecule in normal amounts (dysfunctional form).
Acquired Factor X deficiency may result from warfarin or vitamin K deficiency (in the presence of deficiencies of Factors II, VII, and IX), from liver disease (with deficiencies of other factors synthesized in the liver), with DIC, or as an isolated deficiency in cases of amyloidosis. In amyloidosis, Factor X becomes irreversibly bound to amyloid fibrils in the extracellular space, and is thereby removed from the circulation.
See Table 11–2 for information regarding the laboratory evaluation of patients with Factor X deficiency.
Factor XI deficiency is a commonly encountered disorder. Homozygotes typically have less than 20% of normal Factor XI activity. Heterozygotes have 20% to 70% of normal Factor XI activity. The deficiency in almost all cases appears to be a reduced or absent production of a normal molecule, rather than production of an abnormal or dysfunctional molecule.
The vast majority of the cases of Factor XI deficiency are in people of Jewish descent, particularly those of Ashkenazi origin. The frequency of the homozygous deficient state is 0.2% to 0.3% in the Ashkenazi population, and the frequency of the heterozygous state is extremely high, at approximately 5.5% to 11.0%.
The hemorrhagic tendency is variable for both heterozygotes and homozygotes. Patients with Factor XI levels <15% to 20% uncommonly have spontaneous bleeding but frequently have postoperative bleeding, and patients with levels between 20% and 65% tend to be asymptomatic or have low rates of postoperative bleeding. Bleeding does not correlate well with the level of Factor XI activity. Some homozygotes have an abnormal partial thromboplastin time (PTT), a very low Factor XI level of less than 10%, and no bleeding, even with surgery. The bleeding tendency of a particular individual is more closely related to the bleeding tendency of the patient’s kindred than to the measured Factor XI level. The explanation, which is true for all mutations affecting coagulation factors, is that certain mutations produce a low level of Factor XI and a prolonged PTT but are not clinically significant in vivo. This is because they only affect the activity of the factor in the in vitro clotting factor assays, which are not exact replicas of clot formation in vivo. Acquired decreases in Factor XI can occur with pregnancy, proteinuria, liver dysfunction, and DIC.
See Table 11–2 for information regarding the laboratory evaluation of patients with Factor XI deficiency.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


