|
Typical Clinical Features |
Microscopic Features |
Ancillary Investigations |
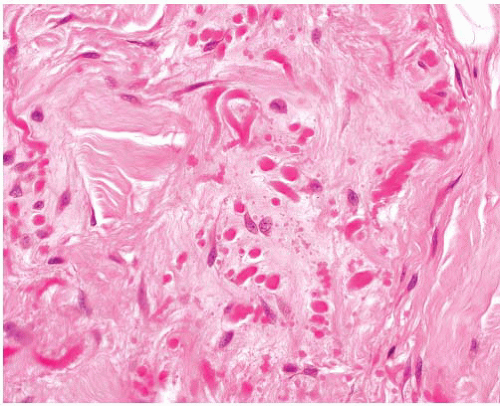
Elastofibroma |
F > M, lower scapular area, beneath bone, infiltrative in subcutis and muscle |
Poorly defined, infiltrative
Thick, focally beaded elastic fibers randomly dispersed in collagen |
Elastic stain positive |
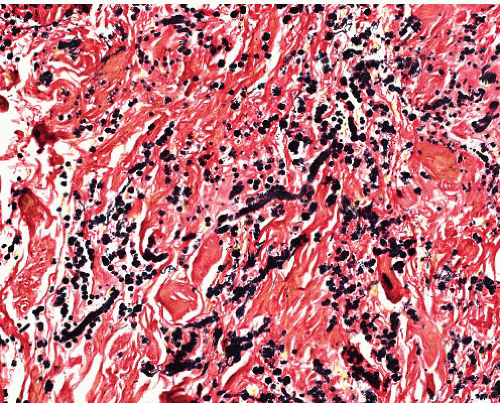
Amyloidoma (tumoral amyloidosis) |
Older adults, extremities, especially lower, in skin or subcutis
Idiopathic, or associated with myeloma, plasmacytoid lymphoma, long-term dialysis, chronic inflammation |
Islands of amorphous eosinophilic material, plasma cells, multinucleated giant cells, calcification, metaplastic bone
Vessel walls involved |
PASD+, apple green birefringence with Congo Red that persists after pretreatment with permanganate in AL amyloid |
Nuchal type and Gardner fibroma |
M > F, neck, shoulder, back
Associated with diabetes and Gardner syndrome
Can later develop desmoid fibromatosis |
Poorly defined infiltrative lesion, dense collagen, very sparse cells, increased entrapped nerves |
CD34+, CD99+
beta-catenin+ |
Nuchal fibrocartilaginous pseudotumor |
Back of neck. History of trauma to neck. At junction of nuchal ligament and deep fascia |
Dense fibrous tissue containing nodules of metaplastic mature cartilage |
S100 protein+ in chondrocytes |
Fibroma of tendon sheath |
M > F, hands, feet, circumscribed
Slowly growing |
Circumscribed, multinodular tumor, comprising hyalinized nodules, spindle cells, slits, or fasciitis-like more cellular areas |
SMA+, CD34-, t(2;11) (q31-32;q12) |
Calcifying fibrous tumor |
Children, young adults, M = F
Subcutaneous, subfascial, or intracavitary |
Circumscribed, unencapsulated lesion
Hypocellular collagen, aggregates of lymphocytes, rounded calcifications, scanty fibroblasts |
Occasionally CD34+, or SMA+ |
Calcifying aponeurotic fibroma |
Childhood, extremities, digits, palm |
Cords and files of spindle or rounded cells in fibrous stroma with focal calcification and chondroid metaplasia
Less commonly, osteoclast-like giant cells and ossification |
S100 protein+ in chondrocytes |
Fibromatosis-superficial |
Palm, sole, penis; infiltrative plaque |
Variable cellular nodules in dense collagen
Cells are parallel aligned, lack atypia
Mast cells |
SMA+, nuclear beta-catenin±, CD34- |
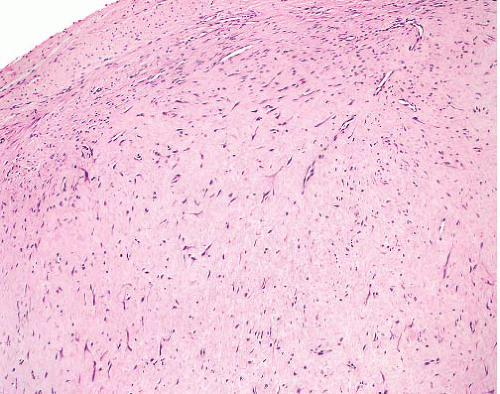
Fibromatosis-desmoid type |
Deep in limbs, head and neck, body cavities |
Parallel myofibroblasts evenly dispersed in collagen, slit-like and thick-walled small-caliber vessels, perivascular and interstitial mast cells
Normal mitoses allowed but no atypia or necrosis |
SMA+, nuclear beta-catenin+, CD34- |
Low-grade fibromyxoid sarcoma |
Skin, deep soft tissue, limbs/girdles |
Fibromatosis-like areas, swirling fibromyxoid transitions, cellular myxoid areas without pleomorphism
Some nuclei lozenge shaped, inconspicuous nucleoli
Giant collagenous rosettes |
Occasionally SMA+, EMA+, claudin-1 in some
Nuclear beta-catenin usually negative t(7;16)(q34;p11), FUS-CREB3L2 or FUSCREB3L1 fusion, rarely EWSR1-CREB3L1 |
Perineurioma |
Skin, subcutis, most locations. Subset in colon
Sclerosing variant: M > F, affects fingers, thumb, palm |
Long thin nuclei, very long slender terminal cytoplasmic processes
Fascicles or perivascular whorls
Fibrous or myxoid stroma
Sclerosing variant has cords and whorls of rounded or epithelioid cells in dense stroma |
EMA+, claudin-1+, CD34+ in some, GLUT-1+, beta-catenin- |
Sclerosing epithelioid fibrosarcoma |
Deep soft tissue, limbs/girdles, head and neck
Can involve or arise in bone |
Multinodular, focally calcified
Cellular islands in dense fibrosis, nests of ovoid cells, clear cytoplasm, or single files simulating carcinoma
Fibrosarcoma-like spindle cell areas in many cases |
Occasional and variable expression of bcl-2, EMA, CK, S100 protein
No specific immunophenotype
EWSR1-CREB3L1 fusions |
Infantile digital (inclusion body) fibromatosis |
Congenital or up to about 2 years, rarely older, F > M |
|
Digits except first
Rarely other sites
Can be multiple
50% of digital lesions recur |
Infiltrative, moderately cellular, bland cells
Rounded eosinophilic paranuclear inclusions |
SMA+, occasionally nuclear beta-catenin+ or CD34+ |
Fibromatosis colli |
Infants in first months, M > F
Mass lower third of sternomastoid or trapezius
Associated with torticollis
Eventual regression |
Infiltrative, at first cellular, later fibrous, between muscle bundles that show focal atrophy or swelling |
Nil specific |
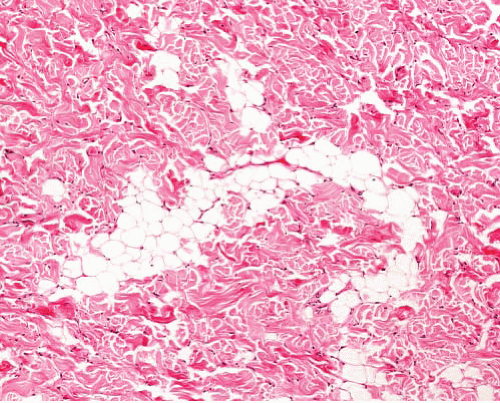
Juvenile hyaline fibromatosis |
Childhood
Skin of head and neck, limbs, bones |
Dense homogeneous eosinophilic stroma with small islands of ovoid cells with clear cytoplasm |
Stroma is positive with PASD and Alcian blue |
Desmoplastic fibroblastoma (collagenous fibroma) |
M > F, mostly subcutaneous, limbs and limb girdles |
Mostly circumscribed, unencapsulated, sparse stellate or spindle cells in dense collagenous stroma, occasional myxoid change, hyalinized vessels |
Focal SMA+, rare nuclear beta-catenin+, t(2;11) (q31;q12) |