KEY POINTS
The incremental interchange of ideas across the specialties of anesthesia and surgery demonstrates the collaborative nature of science in general and medicine in particular. Many surgeons contributed to the growth in anesthesia; more comprehensive anesthesia, in turn, allowed more complex surgery to develop.
The role of the anesthesiologist has expanded to become the perioperative physician. The anesthesiologist evaluates the patient preoperatively, provides the anesthetic, and is involved in postoperative pain relief.
New and improved airway and intubation devices, such as the laryngeal mask airway and the video laryngoscope, along with the American Society of Anesthesiologists’ airway management algorithm, have led to improved management and control of routine and difficult airways.
The specialties of critical care medicine and pain medicine have grown out of the expanded field of anesthesiology. The postanesthesia care unit gave rise to the intensive care unit; the treatment of acute and chronic pain syndromes by anesthesiologists contributed to the growth of pain medicine as a specialty.
The study of proteomics will lead to anesthetics tailored to individuals, maximizing effects and reducing side effects of various anesthetic drugs.
TRUE COLLABORATION
The discipline of anesthesia embodies control of three great concerns of humankind: consciousness, pain, and movement. The field of anesthesiology combines the administration of anesthesia with the perioperative management of the patient’s concerns, pain management, and critical illness. The fields of surgery and anesthesiology are truly collaborative and continue to evolve together, enabling the care of sicker patients and rapid recovery from outpatient and minimally invasive procedures.
BRIEF HISTORY OF ANESTHESIA
The discovery of anesthesia is one of the seminal American contributions to the world. Along with infection control and blood transfusion, anesthesia has enabled surgery to occupy its fundamental place in medicine. Before the advent of modern anesthesia in the 1840s, many substances and methods were tried in the search for pain relief and better operating conditions. Opium, alcohol, exposure to cold, compression of peripheral nerves, constriction of the carotid arteries to produce unconsciousness, and hypnosis (mesmerism) all proved less than satisfactory and dictated rapid and crude surgical procedures. Patients had to be restrained by several attendants, and only the most stoic could tolerate the screams heard in the operating theater. Charles Darwin, who witnessed two such operations, “rushed away before they were completed. Nor did I ever attend again, for hardly any inducement would have been strong enough to make me do so; this being long before the blessed days of chloroform. The two cases fairly haunted me for many a long year.”1
Although Humphrey Davy (1778–1829) suggested using nitrous oxide for the relief of pain in surgical procedures in 1800, this was not pursued until 1844 by dentist Horace Wells (1815–1848). Wells astutely observed that a man who was injured after inhaling nitrous oxide during an exhibition of the “laughing gas”displayed no awareness of pain. After experimenting on himself, Wells attempted to demonstrate the analgesic effects of nitrous oxide for a dental procedure at Harvard Medical School in 1845. The public demonstration was a failure because nitrous oxide has analgesic properties, but does not suffice as the sole anesthetic agent in every patient. Wells never recovered from his humiliating experience and eventually committed suicide. However, he does hold a place in history as the first person to recognize and use the only anesthetic from the 1800s that is still in use today—nitrous oxide.
In 1842, Crawford Long (1815–1878), a physician in rural Georgia, used diethyl ether to induce surgical anesthesia for the removal of two small neck tumors. Diethyl ether had been known for over 800 years but was not used for analgesic purposes. It became an inexpensive and popular recreational drug in the mid-nineteenth century and was used by American medical students at “ether frolics.” Although Long did experiments to verify the analgesic effects of ether, he did not publish his work until 1848, in the Southern Medical Journal, too late to be the unquestioned discoverer of anesthesia.2
William Morton (1819–1868) was a dentist and partner of Horace Wells. After taking a course in anesthesia from Wells, Morton left the partnership in Hartford, Connecticut, and established himself in Boston. He continued his interest in anesthesia, but with diethyl ether replacing nitrous oxide. Ether proved a good choice, as it supports respiration and the cardiovascular system at analgesic levels and is potent enough to administer in room air without hypoxia. He practiced the administration of ether on a dog and then used it when extracting teeth from patients in his office. On October 16, 1846, Morton gave the first public demonstration of ether as an anesthetic for Johns Collins Warren, distinguished surgeon and a founder of Massachusetts General Hospital. In attendance in the surgical amphitheater were several surgeons, medical students, and a newspaper reporter. After anesthesia was induced using a makeshift inhaler, Warren successfully removed a vascular mass from the patient’s neck with no ill effects. Warren was an originator of the Boston Medical and Surgical Journal (now The New England Journal of Medicine), and by November 1846, the demonstration was published in an article by Henry J. Bigelow.3 The stature of Warren and Bigelow lent considerable credence to the advent of surgical anesthesia; as news spread rapidly, surgeons around the world were quick to adopt this “American invention.” Massachusetts General Hospital has restored and preserved the original amphitheater where the demonstration took place, now called the Ether Dome. It is designated as a Registered National Historic Landmark commemorating the first public demonstration, rather than discovery, of the use of ether as an anesthetic.
John Snow (1813–1858) made science out of the art of anesthesia. He was a respected London physician who applied a scholarly, scientific method to investigate the clinical properties and pharmacology of ether, chloroform, and other anesthetic agents. Snow was an astute observer and published a detailed account of the five degrees of etherization in 1847. He vastly improved the apparatus for administering ether and mastered the clinical techniques of anesthetizing patients. As the leading anesthetist of his day, he gave anesthetics to the royal family, including chloroform during labor to Queen Victoria for the birth of Prince Leopold. The Queen’s endorsement of “that blessed chloroform” removed the moral and social stigma against relieving pain during childbirth and brought anesthesia into public awareness. Chloroform, popularized in England by James Simpson (1811–1870), had a narrow therapeutic index and placed great clinical demands on the anesthetist. Ether, with its ability to maintain the cardiovascular and respiratory systems, remained in common use in the United States and often was administered by house staff, medical students, or nurses. Snow encouraged the administration of anesthesia by a physician and felt that a physician dedicated specifically to that purpose was appropriate and necessary. Snow and other exceptional British physicians specializing in anesthesia (Joseph Clover [1825–1882] and Sir Frederick Hewitt [1857–1916]) created a standard of excellence in the latter half of the nineteenth century. This atmosphere of professionalism led to the formation of anesthesia societies and the publication of papers in the prestigious British Medical Journal and The Lancet in England years before such organizations existed in America.4
The ancient Incas chewed coca leaves as a stimulant and may have been aware of its local anesthetic properties, allegedly facilitating trephination of the skull by chewing a clump of coca leaves and dripping the resultant saliva into the wound. The active alkaloid of the coca leaf was synthesized in 1860 and called cocaine by German chemist Albert Niemann, who noted that it “benumbs the nerves of the tongue, depriving it of feeling.”5 Sigmund Freud (1856–1939) of Vienna received a supply of cocaine from Merck, studied its properties, and wrote the famous monograph “Uber Coca” in 1884. Freud was primarily interested in the stimulant and euphoric effects of cocaine and attempted to use it to treat morphine addiction. Freud and Karl Koller (1857–1944), an ophthalmologic intern, began to perform physiologic experiments with cocaine, measuring its effects on muscle strength. Although they both noted that the drug caused numbness of the tongue when swallowed, it was Koller who first instilled it into his own cornea; report of its use as a local anesthetic galvanized the medical world. Soon after, young American surgeons William Halsted (1852–1922) and Richard Hall described intradermal injection of cocaine and were the first to use it for regional blocks of the facial nerves, brachial plexus, and internal pudendal and posterior tibial nerves.6 Halstead later became the first professor of surgery and chief surgeon at Johns Hopkins University, where he remained for more than 30 years. One of the founding fathers of modern surgery, he pioneered radical mastectomy with lymphadenectomy and the use of rubber gloves. While experimenting on themselves, Halstead and other early researchers became addicted to cocaine.7 Its toxic effects were the stimulus to find other local anesthetics—procaine was synthesized in 1905 and lidocaine in 1943.
The New York neurologist Leonard Corning (1855–1923) observed the regional blocks of Halstead and Hall, analytically studied local anesthesia effects on dogs, applied his knowledge to humans, and published the first textbook on local anesthesia in 1886. After experimenting on the spinal nerves of a dog, he intradurally injected a solution of cocaine into a patient, called it spinal anesthesia, and commented that it might be useful in surgery. His suggestion went unheeded for more than 10 years, until August Bier (1861–1949), a prominent German surgeon, gave the first deliberate spinal anesthetic.8 This incremental interchange of ideas and advances across the Atlantic and across the specialties of anesthesia and surgery demonstrates the collaborative nature of science in general and medicine in particular. The development of surgery and anesthesia exemplifies the dichotomy of two fledgling specialties that are mutually dependent, yet increasingly autonomous.
Developments in anesthesia on both sides of the Atlantic progressed rapidly in the twentieth century. The convergence of technologies that produced the hollow needle and syringe, coupled with the synthesis of barbiturates, gave rise to intravenous (IV) anesthesia in the early 1900s. Barbital, followed by hexobarbital and thiopental in 1934, produced rapid and more pleasant induction of anesthesia than the inhaled gases. The concept of “balanced anesthesia” began in 1925, when John Lundy (1894–1973) proposed the use of thiopentone for induction, followed by inhaled agents for maintenance of anesthesia. Lundy directed the department of anesthesiology at the Mayo Clinic for 28 years. He established the first recovery room and blood bank, authored the first textbook on modern anesthesia, and helped found the American Board of Anesthesiology.
Nitrous oxide, diethyl ether, and chloroform, all discovered fortuitously by observation, remained the dominant inhalation agents until the accidental discovery of cyclopropane’s anesthetic properties in 1923. Although rapid acting and pleasant smelling, cyclopropane was limited by its flammability and cardiac irritability. Because it was known that fluorination would reduce or eliminate flammability of chemical compounds, British chemist Charles Suckling set out to synthesize an anesthetic that was stable, potent, volatile, and not flammable. He successfully produced halothane in 1953. Introduced into clinical practice in 1956 after extensive testing in Manchester, England, and paired with an accurate calibrated vaporizer, halothane quickly became the most widely used fluorinated anesthetic. Enflurane and isoflurane, synthesized in the United States by Ross Tyrell, were introduced into clinical practice in 1972 and 1981, respectively. The newest agents, desflurane and sevoflurane, were introduced into clinical practice in the early 1990s. They possess a low solubility and are characterized by rapid onset and recovery, making them particularly well suited to outpatient surgery.
The motto of the American Society of Anesthesiologists (ASA) is “Vigilance,” and to that end, there has been continued progress in objective mechanical measurement of patient well-being. The early anesthesiologists used clinical signs such as patient color, depth of respiration, and pulse rate to monitor depth of anesthesia and patient homeostasis. Harvey Cushing, who eventually became Moseley Professor of Surgery at the Peter Bent Brigham Hospital, began the first anesthesia records or “ether charts” in 1895 while a medical student. They recorded pulse, respiratory rates, pupillary diameter, and the amounts of ether and other drugs administered. He later introduced the use of the portable sphygmomanometer of Riva-Rocci to measure blood pressure and the precordial stethoscope to monitor breath and heart sounds. Monitoring has since progressed to its current state with incremental developments in electrocardiography, pulse oximetry, and mass spectrometry, all mandatory for the safe administration of any anesthetic.
The control of the patient’s airway and respiration as the purview of the anesthesiologist evolved with techniques of endotracheal intubation as pioneered by Sir Ivan Magill (1888–1986) and the invention of the cuffed endotracheal tube by Arthur Guedel (1883–1965). This later merged with the invention of mechanical ventilation and its introduction to the operating room as the embodiment of today’s anesthesia machine. It was this expertise at control of respiration that paved the way for the most revolutionary modern development in anesthesia—the use of muscle relaxants. Curare, a nondepolarizing muscle relaxant, was popularized by Harold Griffith of Montreal. His report of the successful use of curare was a galvanizing event that revolutionized the practice of anesthesia, as the relaxation of abdominal muscles could be controlled to facilitate surgery.9 The depolarizing relaxant succinylcholine was introduced in 1949, and research has continued to provide the newer nondepolarizing drugs mivacurium, pancuronium, rocuronium, atracurium, and cisatracurium.
The specialty of anesthesia is no longer limited to the operating room. It is natural that anesthesiology, born out of the quest to relieve pain, gave rise to the field of acute and chronic pain medicine. The anesthesiologist consulting on the acute pain service may recommend oral, intramuscular, or IV analgesia with a variety of agents, or patient-controlled analgesia. Postsurgical patients also may be treated with nerve blocks: regional (e.g., brachial plexus, popliteal, and femoral) or neuraxial (epidural or intrathecal). The discipline of chronic pain addresses patients who suffer for months or years with cancer or other debilitating diseases. Treatment modalities escalate from orally administered drugs, to diagnostic and therapeutic nerve blocks, to more invasive measures like dorsal column nerve stimulators and radiofrequency or cryosurgical nerve ablation.
Daily management of the airway, fluids and transfusions, ventilation, drug delivery, monitoring, and caring for the sickest patients in the postanesthesia care unit prepared anesthesiologists to become major contributors to the development of critical care medicine. Of the 28 founding members of the Society of Critical Medicine, 10 were anesthesiologists.10
The American Board of Anesthesiology became an independent board in 1941 and, since then, has granted board certification to more than 25,000 diplomates. Certificates in Anesthesia Pain Management and Anesthesia Critical Care Medicine are granted to those completing additional postgraduate training. The ASA has more than 35,000 members, and its official journal, Anesthesiology, has a monthly circulation of 40,000 worldwide.
BASIC PHARMACOLOGY
Pharmacokinetics or the time dependency of a drug describes the relationship between the dose of a drug and its plasma or tissue concentration. It is what the body does to the drug. It relates to absorption, distribution, metabolism, and elimination. The route of administration, metabolism, protein binding, and tissue distribution all affect the pharmacokinetics of a particular drug.
Administration of a drug affects its pharmacokinetics, as there will be different rates of drug entry into the circulation. For example, the oral and IV routes are subject to first-pass effect of the portal circulation; this can be bypassed with the nasal or sublingual route. Other routes of drug administration include transdermal, intramuscular, subcutaneous, or inhalation.
Distribution is the delivery of a drug from the systemic circulation to the tissues. Once a drug has entered the systemic circulation, the rate at which it will enter the tissues depends on several factors:
Molecular size of the drug, capillary permeability, polarity, and lipid solubility. Small molecules will pass more freely and quickly across cell membranes than large ones, but capillary permeability is variable and results in different diffusion rates. Renal glomerular capillaries are permeable to almost all non–protein-bound drugs; capillaries in the brain are fused (i.e., they have tight junctions) and are relatively impermeable to all but the tiniest molecules (the blood-brain barrier). Un-ionized molecules pass more easily across cell membranes than charged molecules; diffusability also increases with increasing lipid solubility.
Plasma protein and tissue binding. Many drugs bind to circulating proteins like albumin, glycoproteins, and globulins. Disease, age, and the presence of other drugs will affect the amount of protein binding; drug distribution is affected because only the unbound free portion of the drug can pass across the cell membrane. Drugs also bind reversibly to body tissues; if they bind with high affinity, they are said to be sequestered in that tissue (e.g., heavy metals are sequestered in bone).11
The fluid volume in which a drug distributes is termed the volume of distribution (Vd). This mathematically derived value gives a rough estimation of the overall physical distribution of a drug in the body. A general rule for volume distribution is that the greater the Vd, the greater the diffusability of the drug. Because drugs have variable ionization rates and bind differently to plasma proteins and tissues, the Vd is not a good predictor of the actual concentration of the drug after administration. Determining the apparent Vd (dose/concentration) is an attempt to more accurately ascertain the drug dose administered and its final concentration.
Metabolism is the permanent breakdown of original compounds into smaller metabolites. Drug elimination varies widely; some drugs are excreted unchanged by the body, some decompose via plasma enzymes, and some are degraded by organ-based enzymes in the liver. Many drugs rely on multiple pathways for elimination (i.e., metabolized by liver enzymes and then excreted by the kidney).
When a drug is given orally, it reaches the liver via the portal circulation and is partially metabolized before reaching the systemic circulation. This is why an oral dose of a drug often must be much higher than an equally effective IV dose. Some drugs (e.g., nitroglycerine) are hydrolyzed presystemically in the gut wall and must be administered sublingually to achieve an effective concentration.
It is important to remember that the response to drugs varies widely. The disposition of drugs is affected by age; weight; sex; pregnancy; disease states; and the concomitant use of alcohol, tobacco, and other licit and illicit drugs. Genetic polymorphism, or variations in genes that cause differing drug effects, is another explanation of varying drug response. This will be discussed later in the Future Direction of Anesthesia section on proteomics. This as yet unpredictable response to drugs underscores the importance of the most important monitor in the operating room—the anesthesiologist, who continuously assesses the patient’s vital signs and adjusts the doses of anesthetic agents to match the surgical stimulus.
Pharmacodynamics, or how the plasma concentration of a drug translates into its effect on the body, depends on biologic variability, receptor physiology, and clinical evaluations of the actual drug. It is what the drug does to the body. An agonist is a drug that causes a response. A full agonist produces the full tissue response, and a partial agonist provokes less than the maximum response induced by a full agonist. An antagonist is a drug that does not provoke a response itself, but blocks agonist-mediated responses. An additive effect means that a second drug acts with the first drug and will produce an effect that is equal to the algebraic summation of both drugs. A synergistic effect means that two drugs interact to produce an effect that is greater than expected from the two drugs’ algebraic summation.12
Hyporeactivity means a larger than expected dose is required to produce a response, and this effect is termed tolerance, desensitization, or tachyphylaxis. Tolerance usually results from chronic drug exposure, either through enzyme induction (e.g., alcohol) or depletion of neurotransmitters (e.g., cocaine).
The potency of a drug is the dose required to produce a given effect, such as pain relief or a change in heart rate. The average sensitivity to a particular drug can be expressed through the calculation of the effective dose; ED50 would have the desired effect in 50% of the general population. The efficacy of any therapeutic agent is its power to produce a desired effect. Two drugs may have the same efficacy but different potencies. The difference in potency of the two drugs is described by the ratio ED50b/ED50a, where a is the less potent drug. If the ED50b equals 4 and the ED50a equals 0.4, then drug a is 10 times as potent as drug b. For example, 10 mg of morphine produces analgesia equal to that of 1 mg of hydromorphone. They are equally effective, but hydromorphone is 10 times as potent as morphine.
Dose-response curves show the relationship between the dose of a drug administered (or the resulting plasma concentration) and the pharmacologic effect of the drug. The pharmacologic effect might be secretion of a hormone, a change in heart rate, or contraction of a muscle. Between 20% and 80% of the maximum effect, the logarithm of the dose and its response has a linear relationship. The term dose only applies to the amount administered and not the actual concentration. If the concentration of an antagonist is increased (in the presence of a fixed concentration of agonist), the dose-response curve will be shifted to the right, and a higher agonist concentration will be required to achieve the desired effect. A basic dose-response curve is shown in Fig. 46-1.
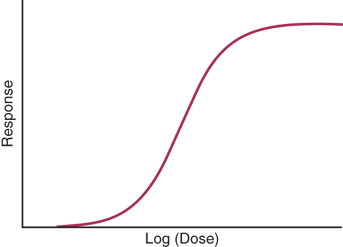
The lethal dose (LD50) of a drug produces death in 50% of animals to which it is given. The ratio of the lethal dose and effective dose, LD50/ED50, is the therapeutic index. A drug with a high therapeutic index is safer than a drug with a low or narrow therapeutic index.
ANESTHETIC AGENTS
Anesthesia can be local, regional, or general (Table 46-1). Local anesthesia is accomplished using a local anesthetic drug that can be injected intradermally and is used for the removal of small lesions or to repair traumatic injuries. Local anesthesia is the most frequent anesthetic administered by surgeons and may be accompanied by IV sedation to improve patient comfort.
| EFFECT | MONITOR | IV DRUGS | POTENT GASES | WEAK GAS | LOCAL ANESTHETICSc | |
|---|---|---|---|---|---|---|
| Unconscious (amnesia) (anxiolysis) | EEG and/or clinical signs | Benzodiazepines Midazolam Lorazepam Diazepam Barbiturates Etomidate Ketaminea | Sevoflurane Desflurane Isoflurane Enflurane Halothane | Nitrous oxideb | ||
| Analgesia | Heart rate Blood pressure Respiratory rate | Opioids Fentanyl Morphine Hydromorphone Nonopioid Ketaminea Parecoxib Dexmedetomidine Acetaminophen IV Ketorolac | Sevoflurane Desflurane Isoflurane Enflurane Halothane | Nitrous oxideb | Amides Lidocaine Bupivacaine Ropivacaine Prilocaine Mepivacaine | Esters Cocaine Procaine Chloroprocaine Tetracaine Benzocaine |
Peripheral nerve blocks (brachial plexus, femoral, etc.) Central nerve blocks (spinal, epidural) | ||||||
| Muscle relaxation (paralysis) | Nerve stimulator | Depolarizer Succinylcholine Nondepolarizers Pancuronium Vecuronium Rocuronium Atracurium | Sevoflurane Desflurane Isoflurane Enflurane Halothane | |||
General anesthesia describes a triad of three major and separate effects: unconsciousness (and amnesia), analgesia, and muscle relaxation (see Table 46-1). IV drugs usually produce a single, discrete effect, while most inhaled anesthetics produce elements of all three. General anesthesia is achieved with a combination of IV and inhaled drugs, each used to its maximum benefit. The science and art of anesthesia are dynamic processes. As the amount of stimulus to the patient changes during surgery, the patient’s vital signs are used as a guide and the quantity of drugs is adjusted, maintaining an equilibrium between stimulus and dose. General anesthesia is what patients commonly think of when they are to be “put under” and can be a cause of considerable preoperative anxiety.13
The IV agents that produce unconsciousness and amnesia are frequently used for the induction of general anesthesia. They include barbiturates, benzodiazepines, propofol, etomidate, and ketamine. Except for ketamine, the following agents have no analgesic properties and do not cause paralysis or muscle relaxation.
The most common barbiturates are thiopental, thiamylal, and methohexital. The mechanism of action is at the γ-aminobutyric acid (GABA) receptor, where they inhibit excitatory synaptic transmission. They produce a rapid, smooth induction within 60 seconds, and wear off in about 5 minutes. In higher doses and in patients with intravascular depletion, they cause hypotension and myocardial depression. The barbiturates are anticonvulsants and protect the brain during neurosurgery by reducing cerebral metabolism.
Propofol is an alkylated phenol that inhibits synaptic transmission through its effects at the GABA receptor. With a short duration, rapid recovery, and low incidence of nausea and vomiting, it has emerged as the agent of choice for ambulatory and minor general surgery. Additionally, propofol has bronchodilatory properties that make its use attractive in asthmatic patients and smokers. Propofol may cause hypotension and should be used cautiously in patients with suspected hypovolemia and/or coronary artery disease (CAD), the latter of which may not tolerate a sudden drop in blood pressure. It can be used as a continuous infusion for sedation in the intensive care unit setting. Propofol is an irritant and frequently causes pain on injection.
The most important uses of the benzodiazepines are for reduction of anxiety and to produce amnesia. Frequently used IV benzodiazepines are diazepam, lorazepam, and midazolam. They all inhibit synaptic transmission at the GABA receptor but have differing durations of action. The benzodiazepines can produce peripheral vasodilatation and hypotension but have minimal effects on respiration when used alone. They must be used with caution when given with opioids; a synergistic reaction causing respiratory depression is common. The benzodiazepines are excellent anticonvulsants and only rarely cause allergic reactions.
Etomidate is an imidazole derivative used for IV induction. Its rapid and almost complete hydrolysis to inactive metabolites results in rapid awakening. Like the above IV agents, etomidate acts on the GABA receptor. It has little effect on cardiac output and heart rate, and induction doses usually produce less reduction in blood pressure than that seen with thiopental or propofol. Etomidate is associated with pain on injection and more nausea and vomiting than thiopental or propofol.
Ketamine differs from the above IV agents in that it produces analgesia as well as amnesia. Its principal action is on the N-methyl-d-aspartate receptor; it has no action on the GABA receptor. It is a dissociative anesthetic, producing a cataleptic gaze with nystagmus. Patients may associate this with delirium and hallucinations while regaining consciousness. The addition of benzodiazepines has been shown to prevent these side effects. Ketamine can increase heart rate and blood pressure, which may cause myocardial ischemia in patients with CAD. Ketamine is useful in acutely hypovolemic patients to maintain blood pressure via sympathetic stimulation but is a direct myocardial depressant in patients who are catecholamine depleted. Ketamine is a bronchodilator, making it useful for asthmatic patients, and rarely is associated with allergic reactions.
The IV analgesics most frequently used in anesthesia today have little effect on consciousness, amnesia, or muscle relaxation. The most important class is the opioids, so called because they were first isolated from opium, with morphine, codeine, meperidine, hydromorphone, and the fentanyl family being the most common. The most important nonopioid analgesics are ketamine (discussed earlier in the Ketamine section) and ketorolac, an IV nonsteroidal anti-inflammatory drug (NSAID).
The commonly used opioids—morphine, codeine, oxymorphone, meperidine, and the fentanyl-based compounds—act centrally on μ-receptors in the brain and spinal cord. The main side effects of opioids are euphoria, sedation, constipation, and respiratory depression, which also are mediated by the same μ-receptors in a dose-dependent fashion. Although opioids have differing potencies required for effective analgesia, equianalgesic doses of opioids result in equal degrees of respiratory depression. Thus, there is no completely safe opioid analgesic. The synthetic opioid fentanyl and its analogues sufentanil, alfentanil, and remifentanil are commonly used in the operating room. They differ pharmacokinetically in their lipid solubility, tissue binding, and elimination profiles and thus have differing potencies and durations of action. Remifentanil is remarkable in that it undergoes rapid hydrolysis that is unaffected by sex, age, weight, or renal or hepatic function, even after prolonged infusion. Recovery is within minutes, but there is little residual postoperative analgesia.
Naloxone and the longer-acting naltrexone are pure opioid antagonists. They can be used to reverse the side effects of opioid overdose (e.g., respiratory depression), but the analgesic effects of the opioid also will be reversed.
Ketamine, an N-methyl-d-aspartate receptor antagonist, is a potent analgesic, but is one of the few IV agents that also causes significant sedation and amnesia. Unlike the μ-receptor agonists, ketamine supports respiration. It can be used in combination with opioids, but the dysphoric effects must be masked with the simultaneous use of sedatives, usually a benzodiazepine like midazolam.
Ketorolac is a parenteral NSAID that produces analgesia by reducing prostaglandin formation via inhibition of the enzyme cyclooxygenase (COX). Intraoperative use of ketorolac reduces postoperative need for opioids. Two forms of COX have been identified: COX-1 is responsible for the synthesis of several prostaglandins as well as prostacyclin, which protects gastric mucosa, and thromboxane, which supports platelet function. COX-2 is induced by inflammatory reactions to produce more prostaglandins. Ketorolac (as well as many oral NSAIDs, aspirin, and indomethacin) inhibits both COX-1 and COX-2, which causes the major side effects of gastric bleeding, platelet dysfunction, and hepatic and renal damage. Parecoxib is a parenteral, predominantly COX-2 NSAID that presumably produces analgesia and reduces inflammation without causing gastrointestinal bleeding or platelet dysfunction.
Dexmedetomidine is an IV α2-adrenergic agonist, administered as a continuous infusion, and has sedative and analgesic properties. It is useful for sedation in an intensive care unit setting and as an adjunct to general anesthesia. Side effects are hypotension and bradycardia.
IV acetaminophen is an analgesic drug and antipyretic of moderate potency; its site of action is in the central nervous system (CNS), not peripherally. It does not have anti-inflammatory properties and is not considered an NSAID.14 When used as part of postoperative analgesic therapy, it will reduce the amount of opioids required, reducing side effects (e.g., constipation, sedation, respiratory depression).
Neuromuscular blocking agents have no amnestic, hypnotic, or analgesic properties; patients must be properly anesthetized before and in addition to the administration of these agents. A paralyzed but unsedated patient will be aware, conscious, and in pain, yet be unable to communicate their predicament. Inappropriate administration of a neuromuscular blocking agent to an awake patient is one of the most traumatic experiences imaginable. Neuromuscular blockade is not a substitute for adequate anesthesia, but is rather an adjunct to the anesthetic. Depth of neuromuscular blockade is best monitored with a nerve stimulator to ensure patient immobility intraoperatively and to confirm a lack of residual paralysis postoperatively.15
Unlike the local anesthetics, which affect the ability of nerves to conduct impulses, the neuromuscular blockers have no effect on either nerves or muscles, but act primarily on the neuromuscular junction.
There is one commonly used depolarizing neuromuscular blocker—succinylcholine. This agent binds to acetylcholine receptors on the postjunctional membrane in the neuromuscular junction and causes depolarization of muscle fibers.
Although the rapid onset (<60 seconds) and rapid offset (5–8 minutes) make succinylcholine ideal for management of the airway in certain situations, total body muscle fasciculations can cause postoperative aches and pains, an elevation in serum potassium levels, and an increase in intraocular and intragastric pressure. Its use in patients with burns or traumatic tissue injuries may result in a high enough rise in serum potassium levels to produce arrhythmias and cardiac arrest. Unlike other neuromuscular blocking agents, the effects of succinylcholine cannot be reversed. Succinylcholine is rapidly hydrolyzed by plasma cholinesterase, also referred to as pseudocholinesterase. There are many reasons for a patient to have low pseudocholinesterase levels, such as liver disease, concomitant use of other drugs, pregnancy, and cancer. These factors are usually not clinically problematic, delaying return of motor function only by several minutes. Some patients have a genetic disorder manifesting as atypical plasma cholinesterase; the atypical enzyme has less-than-normal activity, and/or the patient has extremely low levels of the enzyme. The incidence of the homozygous form is approximately 1 in 3000; the effects of a single dose of succinylcholine may last several hours instead of several minutes. Treatment is to keep the patient sedated and unaware he or she is paralyzed, continue mechanical ventilation, test the return of motor function with a peripheral nerve stimulator, and extubate the patient only after he or she has fully regained motor strength. Two separate blood tests must be drawn: pseudocholinesterase level to determine the amount of enzyme present, and dibucaine number, which indicates the quality of the enzyme. Patients with laboratory-confirmed abnormal pseudocholinesterase levels and/or dibucaine numbers should be counseled to avoid succinylcholine as well as mivacurium, which is also hydrolyzed by pseudocholinesterase. First-degree family members should also be tested. Succinylcholine is the only IV triggering agent of malignant hyperthermia (discussed later in the Malignant Hyperthermia section).
There are several competitive nondepolarizing agents available for clinical use. The longest acting is pancuronium, which is excreted almost completely unchanged by the kidney. Intermediate-duration neuromuscular blockers include vecuronium and rocuronium, which are metabolized by both the kidneys and liver, and atracurium and cisatracurium, which undergo breakdown in plasma known as Hofmann elimination. The agent with shortest duration is mivacurium, the only nondepolarizer that is metabolized by plasma cholinesterase, and like succinylcholine, is subject to the same prolonged blockade in patients with plasma cholinesterase deficiency. All nondepolarizers reversibly bind to the postsynaptic terminal in the neuromuscular junction and prevent acetylcholine from depolarizing the muscle. Muscle blockade occurs without fasciculation and without the subsequent side effects seen with succinylcholine. The most commonly used agents of this type and their advantages and disadvantages are listed in Table 46-2.
| AGENT | DURATION (H) | ADVANTAGES | DISADVANTAGES |
|---|---|---|---|
| Pancuronium | >1 | No histamine release | Tachycardia; slow onset; long duration |
| Vecuronium | <1 | No cardiovascular effects | Intermediate onset |
| Rocuronium | <1 | Fast onset; no cardiovascular effects | — |
| Mivacurium | <1 | Fast onset; short duration & histamine release | — |
The reversal of neuromuscular blockade is not a true reversal of the drug (as with protamine reversal of heparin) but a reversal of the effect of the neuromuscular blockade. Neuromuscular blocking reversal agents, usually neostigmine, edrophonium, or pyridostigmine, increase acetylcholine levels by inhibiting acetylcholinesterase, the enzyme that breaks down acetylcholine. The subsequently increased circulating levels of acetylcholine prevail in the competition for the postsynaptic receptor, and motor function returns. Use of the peripheral nerve stimulator is required to follow depth and reversal of motor blockade, but it is essential to correlate data from the nerve stimulator with clinical signs that indicate return of motor function, including tidal volume, vital capacity, hand grip, and 5-second sustained head lift.
Unlike the IV agents, the inhalational agents provide all three characteristics of general anesthesia: unconsciousness, analgesia, and muscle relaxation. However, it would be impractical to use an inhalation-only technique in larger surgical procedures, because the doses required would cause unacceptable side effects, so IV adjuncts such as opioid analgesics and neuromuscular blockers are added to optimize the anesthetic. All inhaled anesthetics display a dose-dependent reduction in mean arterial blood pressure except for nitrous oxide, which maintains or slightly raises the blood pressure. Nitrous oxide, although not potent enough to use alone, provides partial anesthesia and allows a second agent to be used in smaller doses, reducing side effects.
Minimum alveolar concentration (MAC) is a measure of anesthetic potency. It is the ED50 of an inhaled agent (i.e., the dose required to block a response to a painful stimulus in 50% of subjects). The higher the MAC, the less potent an agent is. The potency and speed of induction of inhaled agents correlate with their lipid solubility, and this is known as the Meyer-Overton rule. Nitrous oxide has a low solubility and is a weak anesthetic agent, but has the most rapid onset and offset. The “potent” gases (e.g., desflurane, sevoflurane, enflurane, and halothane) are more soluble in blood than nitrous oxide and can be given in lower concentrations, but have longer induction and emergence characteristics.
Sevoflurane and desflurane are the two most recently introduced inhalational agents in common use. Because of their relatively lower tissue and blood solubility, induction and recovery are more rapid than with isoflurane or enflurane.
All of the potent inhalational agents (e.g., halothane, isoflurane, enflurane, sevoflurane, and desflurane), as well as the depolarizing agent succinylcholine, are triggering agents for malignant hyperthermia. Table 46-3 lists the advantages and disadvantages of each agent.
| AGENT | MAC (%) | ADVANTAGES | DISADVANTAGES |
|---|---|---|---|
| Nitrous oxide | 105 | Analgesia; minimal cardiac and respiratory depression | Sympathetic stimulation; expansion of closed air space |
| Halothane | 0.75 | Effective in low concentrations; minimal airway irritability; inexpensive | Cardiac depression and arrhythmia hepatic necrosis; slow elimination |
| Enflurane | 1.68 | Muscle relaxation | Strong smell; seizures |
| No effect on cardiac rate or rhythm | |||
| Isoflurane | 1.15 | Muscle relaxation; no effect on cardiac rate or rhythm | Strong smell |
| Desflurane | 6 | Rapid induction and emergence | Coughing; high cost |
| Sevoflurane | 1.71 | Rapid induction and emergence; pleasant smell; ideal for mask induction | High cost; metabolized by liver |
Local anesthetics are divided into two groups based on their chemical structure: the amides and the esters. In general, the amides are metabolized in the liver, and the esters are metabolized by plasma cholinesterases, which yield metabolites with slightly higher allergic potential than the amides (Table 46-4).
| AGENT | EQUIANESTHETIC CONCENTRATION (%) | APPROXIMATE ANESTHETIC DURATION (MIN) | SITE OF METABOLISM |
|---|---|---|---|
| Esters | |||
| Procaine | 2 | 50 | Plasma |
| Chloroprocaine | 2 | 45 | Plasma |
| Tetracaine | 0.25 | 175 | Plasma |
| Amides | |||
| Prilocaine | 1 | 100 | Liver/lung |
| Lidocaine | 1 | 100 | Liver |
| Mepivacaine | 1 | 100 | Liver |
| Bupivacaine | 0.25 | 175 | Liver |
| Ropivacaine | 0.3 | 150 | Liver |
| Etidocaine | 0.25 | 200 | Liver |
Lidocaine, bupivacaine, mepivacaine, prilocaine, and ropivacaine have in common an amide linkage between a benzene ring and a hydrocarbon chain that, in turn, is attached to a tertiary amine. The benzene ring confers lipid solubility for penetration of nerve membranes, and the tertiary amine attached to the hydrocarbon chain makes these local anesthetics water soluble. Lidocaine has a more rapid onset and is shorter acting than bupivacaine; however, both are widely used for tissue infiltration, regional nerve blocks, and spinal and epidural anesthesia. Ropivacaine is the most recently introduced local anesthetic. It is clinically similar to bupivacaine in that it has a slow onset and a long duration, but is less cardiotoxic. All amides are 95% metabolized in the liver, with 5% excreted unchanged by the kidneys.
Cocaine, procaine, chloroprocaine, tetracaine, and benzocaine have an ester linkage in place of the amide linkage mentioned earlier in the Amides section. Unique among local anesthetics, cocaine occurs in nature, was the first used clinically, produces vasoconstriction (making it useful for topical application, e.g., for intranasal surgery), releases norepinephrine from nerve terminals resulting in hypertension, and is highly addictive. Cocaine is a Schedule II drug. Procaine, synthesized in 1905 as a nontoxic substitute for cocaine, has a short duration and is used for infiltration. Tetracaine has a long duration and is useful as a spinal anesthetic for lengthy operations. Benzocaine is for topical use only. The esters are hydrolyzed in the blood by pseudocholinesterase. Some of the metabolites have a greater allergic potential than the metabolites of the amide anesthetics, but true allergies to local anesthetics are rare.
The common characteristic of all local anesthetics is a reversible block of the transmission of neural impulses when placed on or near a nerve membrane. Local anesthetics block nerve conduction by stabilizing sodium channels in their closed state, preventing action potentials from propagating along the nerve. The individual local anesthetic agents have different recovery times based on lipid solubility and tissue binding, but return of neural function is spontaneous as the drug is metabolized or removed from the nerve by the vascular system.
Toxicity of local anesthetics results from absorption into the bloodstream or from inadvertent direct intravascular injection. Toxicity manifests first in the more sensitive CNS and then the cardiovascular system.



