Amyloidosis
Kajsa Affolter, MD
Key Facts
Clinical Issues
SA: Non-pruritic, waxy papules on scalp, neck, and, face with predilection for periorbital area and plaque-like lesions on hands and flexural areas
MA: Pruritic, vaguely defined, rippled brown macules (“hammered brass”), often on back and interscapular region
LA: Small, occasionally pruritic, waxy papules and lichenified plaques, often on extensor surfaces of lower extremities (shins)
NA: Single or numerous large, waxy nodules on lower extremities, face, neck, scalp, or genitalia
Microscopic Pathology
Systemic amyloidosis
Amyloid in dermis, subcutis, and walls of vessels
Macular, lichen, biphasic amyloidosis
Papillary dermis contains small eosinophilic globules of amyloid that can be subtle
Epidermis contains apoptotic bodies and basal vacuolar change
Perivascular chronic inflammatory cell infiltrate is typically present
Nodular amyloidosis
Dermis and subcutis contain large masses of amyloid focused around vessels and adnexa
Monoclonal plasma cells with Russell bodies
Deposition of amyloid light chain (AL) has been shown in many cases
Ancillary Tests
Immunohistochemistry is superior to other ancillary detection methods
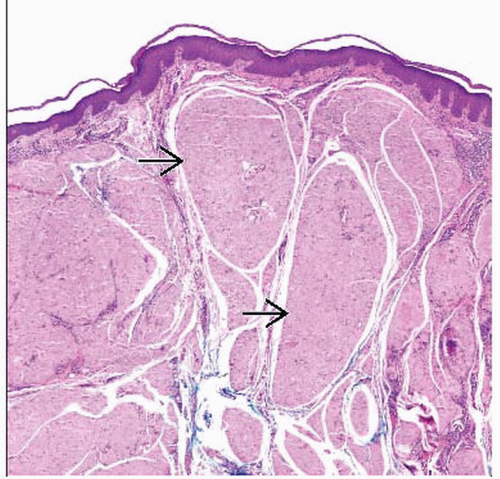 Amyloid is an amorphous, acellular, eosinophilic substance  as seen on H&E section in this case of nodular amyloidosis. as seen on H&E section in this case of nodular amyloidosis. |
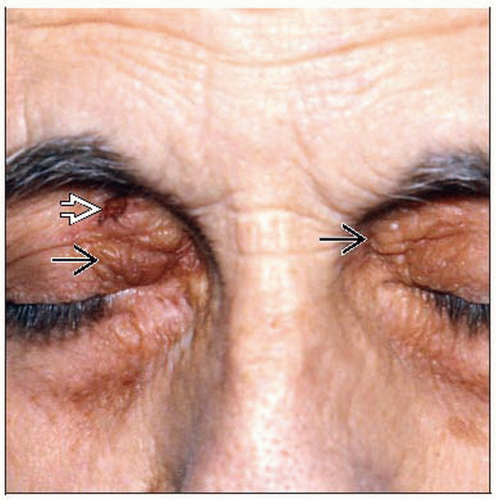 In this image, amyloidosis is demonstrated by waxy indurated papules  on the eyelids. There is also a shave biopsy site on the right upper eyelid on the eyelids. There is also a shave biopsy site on the right upper eyelid  , which confirmed the diagnosis. , which confirmed the diagnosis. |
TERMINOLOGY
Definitions
Generic term denoting extracellular tissue deposition of eosinophilic fibrils composed of altered autologous proteins
ETIOLOGY/PATHOGENESIS
6 Important Amyloid Proteins in Cutaneous Pathology
Amyloid light chain (AL) is derived from immunoglobulin light chains
Amyloid A protein (AA) is an HDL3-associated lipoprotein and an acute phase reactant
Amyloid transthyretin protein (ATTR) is present in certain heritable amyloidosis syndromes
Aβ-2-microglobulin protein induced by β-2-microglobulin glycosylated polypeptide
Amyloid keratin protein (AK) in primary cutaneous amyloidosis derived from filamentous degeneration of keratin filaments
Systemic Amyloidosis (SA)
Primary systemic amyloidosis; often involves skin
Deposition of AL (kappa or lambda)
May be multiple myeloma (MM) associated
Occurs in approximately 1/3 of patients with MM
Secondary systemic amyloidosis
Usually no skin involvement
Deposition of AA, amyloid A, or derived from β-2-microglobulin following long-term hemodialysis
Heritable amyloidosis
Includes entities such as familial Mediterranean fever, Muckle-Wells syndrome, familial cold autoinflammatory syndrome, and familial amyloidotic polyneuropathy
Amyloid elastosis
Rare entity with cutaneous lesions and progressive systemic disease
Primary Localized Cutaneous Amyloidosis (PLCA)
Macular amyloidosis (MA)
Deposition of AK, amyloid keratin protein, which is a keratin intermediate filament
Lichen amyloidosis (LA)
Deposition of amyloid keratin protein (AK), which is a keratin intermediate filament
Nodular amyloidosis (NA)
Deposition of amyloid light chain (AL) has been shown in many cases
Poikilodermatous amyloidosis
Rare form of localized cutaneous amyloidosis
Anosacral amyloidosis
Rare form of localized cutaneous amyloidosis
CLINICAL ISSUES
Epidemiology
Age
PLCA: Predominantly adult population with earliest cases appearing around puberty
Gender
MA: Affects females more than males
LA: Affects genders equally
Ethnicity
SA: In developed countries, AL is most frequently deposited precursor protein, whereas in developing countries AA is most frequently deposited precursor protein
MA is seen with increased frequency in patients from Middle East, Asia, and Central and South America
LA is seen with increased frequency in patients from Southeast Asia
Presentation
Variable depending on organ affected, amount of amyloid deposited, and type of precursor protein
Systemic amyloidosis
Major sites of clinically important involvement include kidneys, heart, skin, and liver
Cutaneous manifestations include
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


