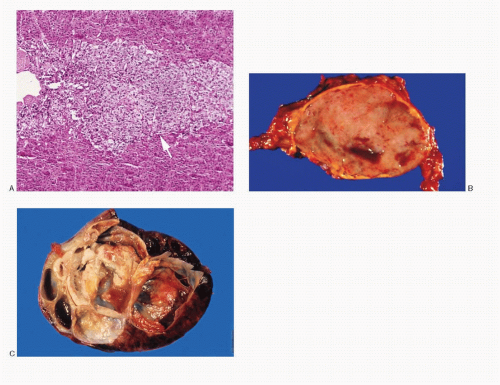
 Fig. 14.1: A: Histologic section of a normal adrenal gland showing external cortex and interior medulla (arrow). B: Gross photograph of a pheochromocytoma. The tumor is well defined, ovoid, and has a smooth cut surface. Note small cystic areas. C: Gross photograph of a different pheochromocytoma with multiple varying-sized cysts, some with hemorrhage. |
giant multinucleated bizarre cells. Stromal amyloid has been reported in 70% of the cases.
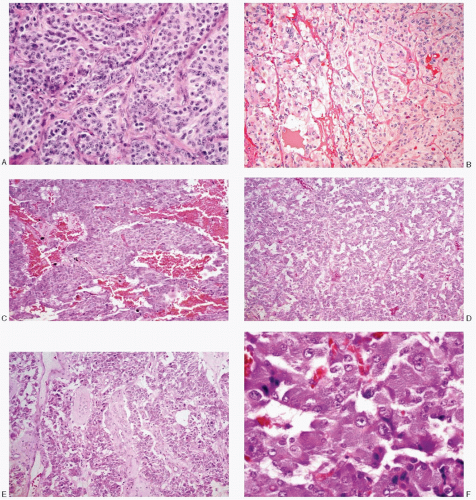 Fig. 14.2: A: Typical alveolar or nesting pattern (Zellballen) formed by large cells, outlined by delicate, vascular stroma. The cells have well-defined borders and appreciable eosinophilic cytoplasm. The nuclei are very uniform with coarsely granular, evenly dispersed chromatin, imparting a salt-pepper appearance. Nucleoli are conspicuous (H&E). B: Another example of an alveolar or nesting pattern. The cells and their nuclei are uniform. There is abundant pale eosinophilic cytoplasm (H&E). C: This pheochromocytoma demonstrates a trabecular pattern with broad trabeculae of uniform medium-sized cells with eosinophilic cytoplasm and high N/C ratios (H&E). D: Pheochromocytoma with a diffuse growth pattern, formed by small round to cuboidal cells with high N/C ratios (H&E). E: Degenerative and cystic changes have resulted in a pseudopapillary pattern (H&E). F: Higher magnification of a histologic section of pheochromocytoma, showing nests of large polygonal cells containing abundant, deep-staining, granular eosinophilic cytoplasm. The nuclei are uniform. The N/C ratios are low (H&E). |
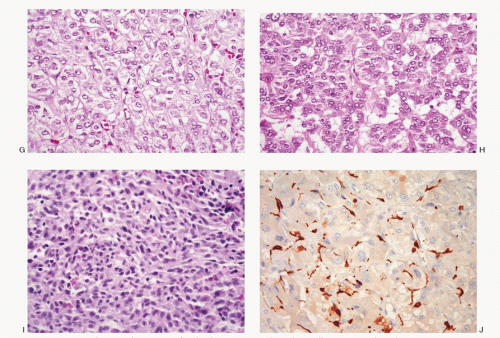 Fig. 14.2: (continued) G: Histologic section of a pheochromocytoma with neoplastic cells containing pale to clear abundant cytoplasm. Note the alveolar pattern (H&E). H: Histologic section of a pheochromocytoma with smaller cells and high N/C ratios (H&E). I: Histologic section of a pheochromocytoma showing a solid growth pattern with small round and spindle cells (H&E). J: Histologic section of a pheochromocytoma, stained with S100 protein, to highlight positively stained sustentacular cells seen at the periphery of the neoplastic cell nests (H&E). |
of giant forms with pyknosis (Figs. 14.6B,C and 14.8C). Intranuclear inclusions may also be present. Their cytoplasm of pheochromocytoma cells is variable. The nuclear/cytoplasmic ratios likewise vary. Some cells appear large and ganglion-like (Fig. 14.8C), while some exhibit wispy cytoplasmic processes (Fig. 14.8B). It is not unusual to encounter disruption of the lipid-rich cytoplasm resulting in a large population of uniform naked nuclei (Figs. 14.12 and 14.13A). The cytoplasm of the pheochromocytoma cells may contain eosinophilic, periodic acid-Schiff (PAS)-positive globules. Red cytoplasmic granules are seen in Romanowsky-stained preparations. Old and new hemorrhage and degenerative changes are frequent.
TABLE 14.1. CYTOPATHOLOGIC FEATURES OF PHEOCHROMOCYTOMA (ADRENAL PARAGANGLIOMA) | ||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


