Key Points
Disease summary:
Hyperammonemia in adults has many different causes. Most are nongenetic. Infrequent but important genetic causes are defects in the urea cycle which are life threatening and treatable.
Urea cycle disorders (UCDs) are caused by defects in the metabolic cycle which converts ammonia to urea.
The adult phenotype may present dramatically or subtly with hyperammonemia of varying degree that can produce cerebral edema. The clinical features include psychosis, altered mental status, vomiting, and focal neurologic signs, or lethargy progressing to obtundation and coma. Acute episodes are often precipitated by surgery, pregnancy, or more likely the postpartum, trauma, and infection.
Chronic features include cognitive and learning deficits, intermittent headaches, intermittent visual disturbances, and focal neurologic signs. There may also be a lifelong aversion to dietary protein.
Hereditary basis:
All the UCDs are inherited in an autosomal recessive manner with the exception of ornithine transcarbamylase (OTC) deficiency which is X-linked.
Differential diagnosis:
Liver failure
Sepsis
Valproate therapy
Diagnostic Criteria and Clinical Characteristics
High ammonia (>100 μmol/L)
Abnormal plasma amino acid profile
Low or high citrulline
Presence of argininosuccinic acid
Low or high arginine
Low or high orotic acid in urine
In the absence of
Liver failure
Overwhelming sepsis
Metabolic acidosis
UCDs interrupt the conversion of ammonia to urea (Fig. 91-1). Ammonia is generated as a by-product of protein catabolism and elevated ammonia levels (>100 μmol/L) appear to be extremely toxic to the central nervous system. UCD in adults can present with chronic symptoms of intermittent headaches, intermittent visual disturbances, cognitive and learning deficits, and focal neurologic signs. These adults have self-selective dietary protein aversion. Acute episodes can be triggered by increased dietary intake of protein or secondary to increased protein breakdown related to catabolic states, such as infection, trauma, surgery, pregnancy, or the postpartum. These symptoms may resolve spontaneously or may progress to hyperammonemic encephalopathy. Patients may present acutely with vomiting, anorexia, lethargy, altered mental status, seizures, focal neurologic signs, or psychosis. Hepatomegaly may be present on clinical examination. Hyperammonemia can produce cerebral edema and patients, if untreated, continue to deteriorate, become comatose, and may die from cerebral herniation. Upon recovery, patients may be left with significant neurologic deficits.
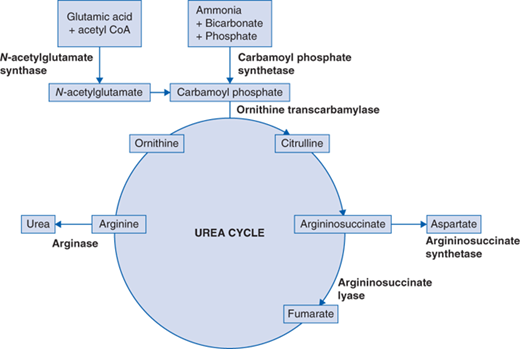
While the urea cycle disorders usually present in infancy or childhood, often dramatically, all have a late-onset form that presents in adulthood with hyperammonemia and is related to partial (hypomorphic) rather than complete enzyme deficiencies. Due to random X-chromosome inactivation, approximately 15% of female carriers with a defect in OTC develop hyperammonemia. Arginase (ARG) deficiency typically presents as a chronic neurologic problem with cognitive reduction and spastic diplegia. Unlike the dramatic clinical presentation of the other urea cycle disorders, patients with argininosuccinate lyase (ASL) deficiency can develop trichorrhexis nodosa (or brittle hair) and often have chronic liver disease.
The most frequent cause of hyperammonemia in adults is acute and/or chronic hepatic failure. Other causes of hyperammonemia include certain drugs, notably valproic acid, and urinary tract infection with a urease-producing organism such as Proteus mirabilis or Klebsiella pneumoniae. Certain chemotherapeutic agents such as 5-fluorouracil, cyclophosphamide, and asparaginase have been shown to reduce urea cycle function and cause hyperammonemia. Rare causes of hyperammonemia in adults include Hashimoto thyroiditis and multiple myeloma.
Screening and Counseling
In evaluating an adult with hyperammonemia, it is important to exclude nongenetic causes such as liver failure or sepsis. First-line investigations include
Ammonia (blood in sodium heparin tube sent STAT to laboratory on ice)
Liver function tests
Electrolytes and blood gases
Plasma amino acids
Urine orotic acid
Note: Ammonia can be spuriously elevated if there is delay in sample analysis. This is the most frequent cause of hyperammonemia in a relatively well individual.
UCDs presenting as hyperammonemic encephalopathy are clinically indistinguishable from each other (Table 91-1). Plasma amino acids are very important in differentiating the various enzyme deficiencies, as highlighted in the algorithm (Fig. 91-2). As depicted in the pathway (Fig. 91-1), the UCDs may be divided into two categories, proximal and distal, depending on the level of citrulline. Low citrulline levels point to the possibility of a proximal disorder like N-acetylglutamate synthase (NAGS), carbamoyl phosphate synthetase (CPS1) or OTC deficiency. Orotic acid, an intermediate in the pyrimidine biosynthetic pathway, is elevated in OTC deficiency but normal or perhaps low in CPS1 and NAGS deficiency. CPS1 and NAGS deficiency are biochemically indistinguishable and require measurement of enzyme activity or molecular genetic testing. Significantly elevated levels of citrulline, argininosuccinic acid, and arginine are suggestive of a distal disorder such as argininosuccinate synthetase (ASS) deficiency, argininosuccinate lyase (ASL) deficiency, or arginase (ARG) deficiency, respectively.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


