Key Points
Disease summary:
Fabry disease is an X-linked lysosomal storage disease (LSD), which results from the deficient activity of the enzyme, α-galactosidase A (α-Gal A), and the lysosomal accumulation of glycosphingolipids with α-terminal galactosyl moieties, primarily globotriaosylceramide (GL-3).
Two major clinical subtypes have been described. The classic phenotype due to mutations that express little, if any, α-Gal A activity (<1% of mean normal) and present in childhood or adolescence, and a later-onset phenotype due to mutations that express residual enzyme activity (>1% of mean normal levels).
Newborn screening studies indicate the incidence in males of the classic phenotype to be about 1 in 25,000 to 40,000, while the later-onset phenotype is about 10 times more frequent in Europe and Taiwan.
Clinical manifestations of the classic phenotype include angiokeratoma, acroparesthesias, hypohidrosis, gastrointestinal symptoms, and a characteristic corneal keratopathy. With advancing age, the classic patients develop renal failure, cardiac disease, cerebrovascular disease, and premature demise.
Patients with the later-onset phenotype do not have endothelial glycosphingolipid deposition and lack the early manifestations of the classic phenotype. They develop renal failure, cardiac disease, or strokes later in life, typically between the third and fifth decades of life.
Enzyme replacement therapy (ERT) with recombinant α-Gal A has proven safe and effective in clinical trials.
Hereditary basis:
Fabry disease (both subtypes) is inherited as an X-linked trait. Manifestations in heterozygous females can range from as severely affected as males to asymptomatic, primarily due to random X-chromosomal inactivation. All daughters of an affected male are heterozygous, while all sons will not inherit the Fabry gene. On average, 50% of sons of a heterozygous mother will be affected and 50% of daughters will be heterozygotes.
Differential diagnosis:
Acroparesthesias:
Many of the symptoms in Fabry disease can be similar to those of other disorders, including rheumatoid or juvenile arthritis, rheumatic fever, erythromelalgia, lupus erythematosus, Raynaud syndrome, fibromyalgia, and multiple sclerosis. In children with acroparesthesias without any other major finding, “growing pains” may be attributed to their complaints.
Angiokeratoma:
Angiokeratomas, that are similar to or indistinguishable from the clinical appearance and distribution of the cutaneous lesions in Fabry disease, have been described in other lysosomal storage diseases, including fucosidosis (OMIM #230000), galactosialidosis (OMIM #256540), GM1-gangliosidosis (OMIM #230500), aspartylglucosaminuria (OMIM #208400), β-mannosidosis (OMIM #248510), and Kanzaki disease (OMIM #609242).
Angiokeratoma of Fordyce, angiokeratoma of Mibelli, and angiokeratoma circumscriptum are a few cutaneous disorders that have similar cutaneous lesions to those seen in Fabry disease. None have the typical histologic or ultrastructural lysosomal pathology of the Fabry lesion.
The angiokeratoma of Fordyce is similar in appearance to that of Fabry disease, but is limited to the scrotum, and usually appears after age 30.
The angiokeratomas of Mibelli includes warty lesions on the extensor surfaces of extremities in young adults and is associated with chilblains.
Angiokeratoma circumscriptum or naeviformus can occur anywhere on the body, is clinically and histologically similar to that of Fordyce, and is not associated with chilblains.
In adults, presence of arrhythmias, left ventricular hypertrophy (LVH), short PR interval, hypertrophic cardiomyopathy, proteinuria, renal insufficiency, transient ischemic attacks (TIAs), and strokes can all be consistent with Fabry disease. One should be on alert especially if there is a family history of these, and an X-linked inheritance pattern.
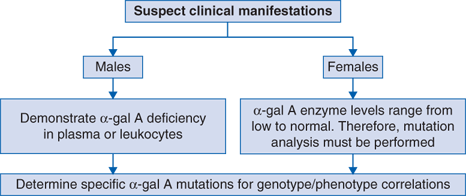
Male patients in hemodialysis or renal transplantation clinics, as well as in cardiac clinics and in stroke clinics, should be evaluated for unrecognized Fabry disease by demonstrating deficient α-Gal A activity in plasma or leukocytes. These results are diagnostic in males, but α-Gal A mutation analysis should be performed to identify the family mutation and for the diagnosis of heterozygotes for which the enzyme assay is unreliable due to random X-chromosomal inactivation.
Diagnostic Criteria and Clinical Characteristics
The major early manifestations in classically affected males and some heterozygous females include acroparesthesias, angiokeratoma, hypohidrosis, and a characteristic corneal keratopathy. With advancing age, the glycosphingolipid (GL-3) deposition in the microvascular endothelial cells throughout the body leads to renal insufficiency and failure, cardiac and cerebrovascular events, and premature demise.
Classically affected patients typically present with neuropathic pain in childhood or adolescence, which also can be misdiagnosed as erythromelalgia or fibromyalgia. Episodes of burning or tingling pain in the hands and feet (acroparesthesias) in children or adolescents are brought on by exercise, fever, stress, or changes in weather conditions. The pain may be chronic, or can be severe and last for days or even weeks.
The presence of cutaneous lesions, termed angiokeratomas, which are actually telangiectasias, in affected males, sparse lesions appear in childhood or adolescence in the “swimsuit” region, especially in the umbilicus and on the scrotum. Heterozygous females from families with the classic phenotype may have sparse angiokeratomas on the breasts, flanks, and/or genital region.
Decreased or absent ability to sweat, hypohidrosis or anhidrosis, is characteristic in classically affected males and many heterozygotes.
The bilateral corneal dystrophy can be mimicked by use of certain drugs including amiodarone and chloroquine.
With advancing age, classically affected males will develop renal insufficiency leading to dialysis and/or transplantation, cardiac disease including left ventricular hypertrophy leading to hypertrophic cardiomyopathy, arrhythmias, and cerebrovascular disease including TIAs and strokes.
Prior to ERT, the average age of death prior to dialysis and renal transplant was about 40 years, with renal treatment, about 50 years in classically affected males.
Males with the later-onset phenotype have residual α-Gal A activity and lack the early manifestations in classically affected males, including the acroparesthesias, angiokeratoma, hypohidrosis, and the corneal keratopathy. They present later in life (third-eighth decades), with renal, cardiac, and/or cerebrovascular disease.
| System | Manifestation |
|---|---|
| Skeletal | Mild, if any, skeletal involvement. Mild osteopenia in older patients |
| Visceral | Hypohidrosis in classically affected males from early childhood |
| Hematologic | Mild anemia in some patients |
| Pulmonary | Glycolipid storage in lungs can cause lung disease. Patients should not smoke |
| Immunologic | N/A |
| Metabolic | The deficient activity of α-galactosidase A results in the systemic accumulation of the glycosphingolipid, globotriaosylceramide (GL-3), and related glycolipids, particularly in the microvascular endothelium |
| Neurologic | In childhood, burning, tingling pain in the extremities. In adults, TIAs and strokes can occur |
| Cardiovascular | Cardiovascular dysfunction may include left ventricular hypertrophy leading to hypertrophic cardiomyopathy, valvular abnormalities, and arrhythmias |
| Ophthalmologic | Characteristic corneal dystrophy on slit-lamp microscopy. Vision is not impaired. Tortuous conjunctival and retinal vessels, characteristic anterior and posterior cataracts |
| Renal | Glomerular involvement leads to renal insufficiency and failure |
| Dermatologic | Classically affected patients have diffuse angiokeratoma (telangiectasias) and hypohidrosis |
| Gastrointestinal | Classically affected males may have chronic postprandial pain and diarrhea |
(Fig. 88-1):
Suspect clinical manifestations include the following in classically affected males: childhood or adolescent onset of acroparesthesias: pain in the fingers and toes, particularly during a fever, exercise, or stress. Typically these males also will have hypohidrosis, angiokeratomas in the swimsuit region, gastrointestinal manifestations (postprandial pain and diarrhea), and a characteristic corneal dystrophy only visible by slit-lamp microscopy. Later-onset patients present in the third to eight decade with renal insufficiency and failure, left ventricular hypertrophy, hypertrophic cardiomyopathy, and/or transient ischemic attacks or cryptogenic strokes.
Demonstration of deficient plasma or leukocyte α-Gal A activity in affected males. Confirm α-Gal A enzyme deficiency by α-Gal A mutation analysis to determine family mutations. Mutation analysis also provides genotype or phenotype correlation for classic and later-onset disease.
For heterozygous females, the α-Gal A activity can range from low to normal and therefore, all suspect heterozygotes are diagnosed by demonstration of the family mutation, which also provides genotype or phenotype information.
Over 670 α-Gal A mutations are known as of August, 2013.
There are two major subtypes of Fabry disease, the classic and later-onset phenotypes. Affected males with the classic phenotype have little, if any, α-Gal A enzyme activity (<1% of mean normal), whereas males with the later-onset phenotype have residual enzymatic activity, typically greater than 1% of normal. In classically affected males, the progressive microvascular endothelial glycosphingolipid accumulation leads to the clinical manifestations including angiokeratomas, acroparesthesias, hypohidrosis, and gastrointestinal abnormalities. They also have a characteristic corneal opacity detectable early in childhood or adolescence. With advancing age, manifestations include renal insufficiency and failure, cardiac involvement, and cerebrovascular disease. Heterozygous females from Fabry families with the classic phenotype have a wide range of clinical manifestations that vary from asymptomatic to severely affected.
Affected males with the later-onset phenotype present in adulthood, usually lack the classical early manifestations (angiokeratomas, acroparesthesias, hypohidrosis, and corneal opacities), and develop renal and/or cardiac disease in the third to fifth decades of life. Heterozygous females from later-onset families may be asymptomatic or develop symptoms later in life, including cardiac and renal manifestations.
The symptoms of the classic subtype of Fabry disease usually begin in childhood or adolescence. Early symptoms include the onset of acroparesthesias and the appearance of angiokeratomas. Pain is an early symptom of the classic phenotype. Affected individuals may experience episodes of severe burning pain in the hands and the feet (acroparesthesia) and sometimes in the arms and legs. Severe episodes of pain (Fabry crises) may last for hours to days and are frequently associated with exercise, fatigue, and/or fever. Classically affected individuals have decreased or absent sweat production (hypohidrosis or anhidrosis) and discomfort in warm temperatures (heat intolerance). The angiokeratomas are cutaneous macular-papular lesions that are reddish to dark-blue and are typically found in the area between the hips and the knees.
Classically affected males and about 90% of heterozygotes from classically affected families have a characteristic corneal opacity seen by slit-lamp microscopy that does not affect vision. Conjunctival and retinal vessels are tortuous and may appear cork screw like and/or dilated due to the glycosphingolipid accumulation in the vessel walls. Additional symptoms associated with Fabry disease may include dizziness, headache, generalized weakness, and fatigue.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


