Key Points
Disease summary
Hereditary angioedema (HAE) is a disorder characterized by episodic local subcutaneous and submucosal edema that typically involves the gastrointestinal and upper respiratory tracts.
There are three clinically indistinguishable types of the disorder. In HAE type I, serum levels of C1 esterase inhibitor (C1NH) are less than 35% of normal. In HAE type II, the levels are normal or elevated, but the protein is nonfunctional. HAE type III occurs exclusively in women. Both concentration and function of C1 inhibitor are normal and are precipitated or worsened by high estrogen levels.
The hereditary form accounts for approximately 2% of clinical angioedema cases and occurs in approximately 1 in 50,000 persons. It affects whites, African Americans, and all other ethnic groups.
Hereditary basis:
Inheritance: autosomal dominant
Diagnostic Criteria and Clinical Characteristics
The diagnosis is established in patients with one clinical criterion and one laboratory criterion. This is true for acquired disorders as well.
Self-limiting, noninflammatory subcutaneous angioedema without urticaria, recurrent, and lasting more than 12 hours.
Self-remitting, recurrent abdominal pain lasting more than 6 hours without clear organic etiology.
Recurrent laryngeal edema.
A family history of recurrent angioedema and/or abdominal pain and/or laryngeal edema, if present, supports the diagnosis of HAE, although it is not required because the patient may have a new mutation or an acquired disorder.
C1 inhibitor levels less than 50% of the lower limit of normal at two separate determinations (at least 1 month apart) with the patient in their basal condition and after the first year of life.
C1 inhibitor function of less than 50% of normal at two separate determinations (at least 1 month apart) with the patient in their basal condition and after the first year of life.
Mutation in C1 inhibitor gene altering protein synthesis and/or function. This is the only laboratory criterion that can be used to make the diagnosis in patients younger than 1 year of age.
The criteria stipulate that C1 inhibitor antigenic levels and functional levels must be below 50%.
The following diagnostic algorithm (Fig. 79-1) was presented at an international consensus conference (Bowen et al, 2004).
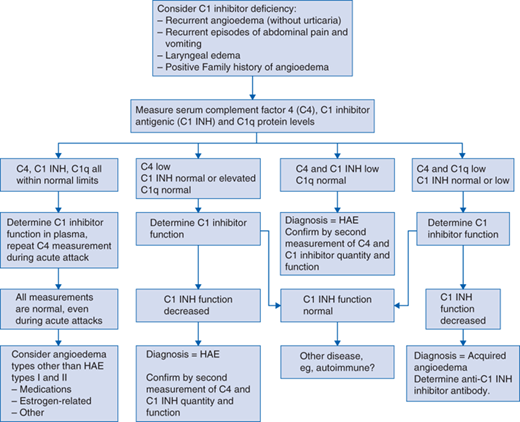
Cutaneous eruptions are characterized by recurrent, nonpruritic angioedema in the absence of hives. Swelling can affect any area of the body, including the extremities, face, trunk, gastrointestinal tract, genitourinary regions, or upper airways. Abdominal symptoms include nausea, vomiting, abdominal pains, and diarrhea and may mimic infantile colic, acute appendicitis, or acute abdomen. Age of onset is variable, and the patient might present at less than 1 year of age with colic. Most patients experience their first attack by age 15, but the diagnosis may also be suspected in young adults, especially in retrospect of earlier symptoms. Although laryngeal symptoms may occur in early childhood, this tends to occur later. Attacks tend to last within 72 hours and usually are periodic, often followed by several weeks of remission.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


