Key Points
Disease summary:
Autoinflammatory diseases (AIDs) are illnesses of the innate immune system without high-titer autoantibodies or antigen-specific T cells in contrast to autoimmune diseases that relate to a deficit of acquired immunity. These diseases are characterized by recurrent fever and systemic inflammation, and the main complication is the risk of generalized or renal amyloidosis in untreated patients. Other sporadic, undefined, or complex disorders have been also linked to the AIDs group. This review will focus on hereditary recurrent fevers (HRFs).
Hereditary basis:
Both dominant and recessive autosomal transmission, with incomplete penetrance
Differential diagnosis:
Nonhereditary recurrent fevers, for example, PFAPA syndrome (Marshall syndrome)
Diagnostic Criteria and Clinical Characteristics
Example: at least one of the following
Recurrent fever accompanied with various association of osteoarticular, cutaneous, and abdominal signs
Biologic marker of inflammation (enhanced C-reactive protein [CRP]) during acute episodes
Familial history
And the absence of
Infection, antibodies
Symptom-free periods, pediatric onset
Screening and Counseling
See Fig. 77-1.
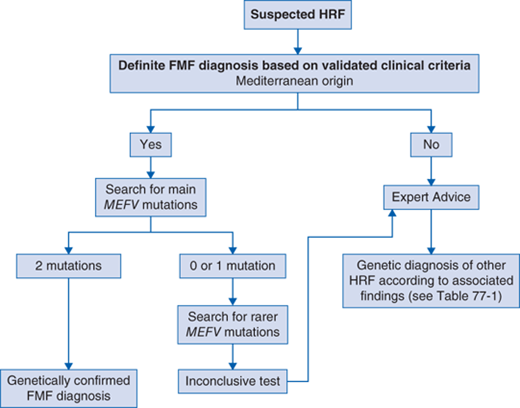
FMF and MKD are recessively inherited. TRAPS and CAPS are dominant diseases, but de novo mutations have been identified in rare cases of TRAPS and in all cases of severe forms of CAPS (also named, chronic infantile neurologic cutaneous and articular syndrome [CINCA]). Because of clinical similarities across AIDs, differential diagnosis is sometimes difficult. A decade ago, specific diagnosis has become possible after the discovery of the respective genes responsible for HRF (Table 77-1). The definitive genetic diagnosis of hereditary AIDs lies on the finding of unambiguous mutations in the causative genes, for example, M694V in the FMF gene, and C30R in the TRAPS gene. However, several variants are currently debated as to whether they are low-penetrance causing mutations or functional polymorphisms. Well-known examples are E148Q for MEFV, R121Q or P75L (old names R92Q and P46L) for TNFRSF1A, and V198M for NLRP3, respectively. A registry of mutations named infevers is available online at http://fmf.igh.cnrs.fr/ISSAID/infevers.
| Syndrome | Gene Symbol | Associated Findings |
|---|---|---|
| FMF: familial Mediterranean fever | MEFV (Mediterranean fever) | Recurrent fever lasting 1-3 days, almost exclusively patients of Mediterranean ancestry, peritonitis, monoarthralgia of the lower extremities, erysipelas-like erythema, splenomegaly |
| MKD: mevalonate kinase deficiency | MVK (mevalonate kinase) | Recurrent fever lasting 3-7 days, diarrhea, cervical adenopathies, acute abdomen bipolar aphtosis, arthralgia, maculopapular rash |
| TRAPS: TNF receptor-associated periodic syndrome | TNFRSF1A (tumor necrosis factor receptor superfamily 1A) | Recurrent fever lasting a few days to a few weeks, cervical stiffness, migratory erythema and myalgia, arthralgia |
| CAPS: cryopyrin-associated periodic syndrome | NLRP3 (NLR family, pyrin domain containing 3) | Recurrent fever lasting a few hours to a few days, circadian profile, urticaria, arthralgia, and in severe cases, amyloidosis, deafness, blindness, bone deformation, meningitis |
The homozygote M694V genotype is associated with earlier onset and more frequent renal amyloidosis, but environmental factors may play a primary role overwhelming that of MEFV
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


