Key Points
Disease summary:
Kallmann syndrome (KS; hypogonadotropic hypogonadism and anosmia) and normosmic idiopathic hypogonadotropic hypogonadism (nIHH) represent the two major clinical presentations of humans with isolated deficiency of gonadotropin-releasing hormone (GnRH).
Both KS and nIHH are more common in males than females (~3:1) and affected patients present with either complete or partial absence of puberty.
During development, GnRH neurons arise from the embryonic olfactory placode and migrate in utero into the mediobasal hypothalamus where they secrete GnRH in a pulsatile manner. Combined developmental failure of both olfactory and GnRH neuronal migration result in KS, while isolated neuroendocrine failure of anatomically normal GnRH neurons results in nIHH.
Hereditary basis:
70% of KS or nIHH cases are apparently sporadic while the remainder is familial. However, on more detailed family expansion, a familial pattern can become evident. The inheritance pattern is heterogeneous. X-linked recessive, autosomal dominant, autosomal recessive, and oligogenic modes of inheritance are all documented.
Differential diagnosis:
In adolescents presenting with delayed puberty, the main differential diagnosis is constitutional delay in puberty (CDP). Time is the major factor in distinguishing CDP and KS or nIHH as subjects with CDP eventually enter puberty spontaneously and achieve normal sexual maturation.
Other anatomic (CNS or pituitary tumors, head trauma, CNS irradiation, etc) or functional (chronic systemic illness, eating disorders, malnutrition) causes of hypogonadotropic hypogonadism must be excluded.
Diagnostic Criteria and Clinical Characteristics
All of the following
Absent or incomplete puberty by age 18 years
Serum testosterone less than or equal to 100 ng/dL in males or serum estradiol (E2) less than or equal to 20 pg/mL in females in the presence of inappropriately low or normal levels of pituitary gonadotropins (luteinizing hormone [LH] and follicle-stimulating hormone [FSH])
All other anterior pituitary hormonal functions are normal and normal anatomy of the hypothalamus and pituitary fossa by magnetic resonance imaging (MRI)
And the absence of
Functional etiology of hypogonadotropic hypogonadism (chronic systemic illness, eating disorders, malnutrition, chronic opiate ingestion, chronic glucocorticoid use, anabolic steroid use)
Evidence of iron overload (hemochromatosis)
Clinical manifestations vary with timing of onset of GnRH deficiency.
Microphallus and cryptorchidism can be seen in infants with neonatal onset of GnRH deficiency and may prompt early diagnosis. Subjects with TAC3/TACR3 signaling pathway mutations tend to exhibit a higher incidence of such neonatal defects.
KS is characterized by the combination of complete anosmia or hyposmia occurring with isolated GnRH deficiency and accounts for approximately 50% of subjects with isolated GnRH deficiency. Anosmia may also be recognized during early childhood prompting early assessment.
The majority of KS and nIHH patients typically present in adolescence with completely absent or arrested early puberty. The remaining subjects present with incomplete puberty, including a characteristic “fertile eunuch” variant in male subjects wherein testicular development and spermatogenesis occur despite overt biochemical hypogonadism.
An extremely rare presentation of isolated GnRH deficiency in males is the adult-onset variant, wherein patients who had otherwise completed normal puberty present with diagnostic criteria for GnRH deficiency later in adulthood.
In approximately 10% KS or nIHH subjects, spontaneous reversal of GnRH deficiency may occur upon hormonal treatment. All KS or nIHH individuals should therefore be screened for spontaneous reversal by periodic withdrawal of hormonal therapy.
Screening and Counseling
At present, disease-causing mutations in 22 different causative genes have been identified in approximately 50% of subjects with isolated GnRH deficiency. A suggested algorithm for screening for mutations in the known genes using our current understanding of the phenotype-genotype correlations is shown in Fig. 67-1.
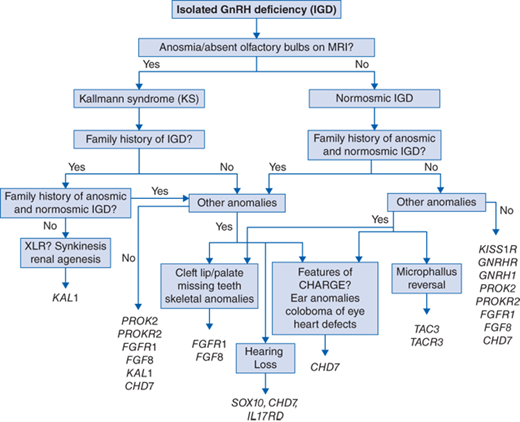
A detailed three-generation family history is a critical initial step for targeted genetic screening and predicting risk in family members. The evolving genetic architecture of human GnRH deficiency poses a significant genetic counseling challenge. Inheritance patterns vary depending on the causative gene and include X-linked recessive (KAL1), autosomal recessive (GNRH1, GNRHR, KISS, KISS1R, TAC3, TACR3), and autosomal dominant (FGFR1, CHD7, SOX10) patterns. Indeterminate inheritance patterns, overlap or mixed pedigrees (ie, KS and nIHH within a single pedigree), pedigrees with incomplete penetrance or variable expressivity warrant specific screening for the KS or nIHH overlap genes (PROK2/PROKR2/FGF8/FGFR1/HS6ST1, FGF17, SPRY4, DUSP6).
Genotype-Phenotype Correlation
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


