Key Points
Disease summary:
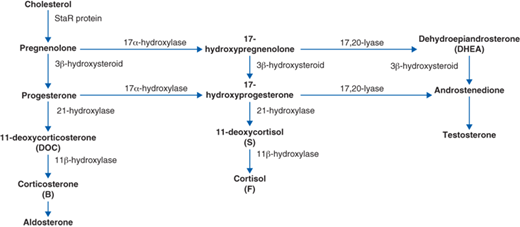
Congenital adrenal hyperplasia (CAH) is a group of autosomal recessive inherited disorders of steroidal biosynthesis caused by a variety of enzymatic defects (Fig. 64-1).
Most cases of CAH can be accounted for by deficiencies in 21-hydroxylase (21-GHO): 90% to 95% of cases and 11β-hydroxylase (OHO): 3% to 8%.
Deficiencies in 21-hydroxylase and 11β-hydroxylase cause decreased cortisol production, that lead to a lack of negative inhibition of adrenocorticotropic hormone (ACTH) and oversecretion of ACTH. This increase in ACTH drives the adrenal glands to attempt to produce more cortisol but this increase is blocked by enzyme deficiencies. Adrenal precursors are thus shunted into the androgen pathway resulting in increased androgen synthesis, which does not require these enzymes.
Phenotypes can range widely depending on the degree of enzyme deficiency.
Hyperandrogenism is a key feature seen in 21-OHD and 11β-OHD.
Hereditary basis:
These are autosomal recessive genetic disorders; thus both parents are typically carriers of CYP21A2 or affected with 21-hydroxylase deficiency for the fetus to be affected.
Though less common, new mutations can arise in CYP21A2.
Differential diagnosis:
In 46,XX, maternal androgen exposure, P450 oxoreductase deficiency (POR), ovotesticular disorder of sexual differentiation (DSD), mixed gonadal dysgenesis
In 46,XY, incomplete androgenization of genitals, 5α-reductase deficiency, nonclassic StAR
Diagnostic Criteria and Clinical Characteristics
Diagnostic evaluation should include
Newborn screen for 21-hydroxylase deficiency in all children born in the United States.
All patients born with ambiguous genitalia must have a detailed hormonal and genetic evaluation.
Genotyping is necessary as each mutation can cause a different phenotype.
Hormonal determination
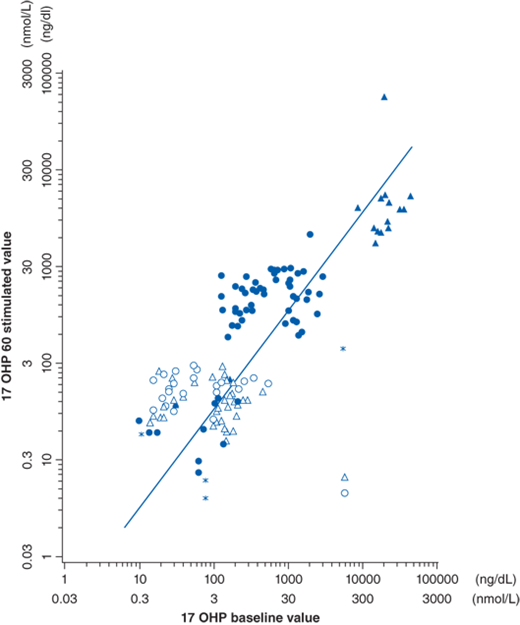
The gold standard for establishing hormonal diagnosis of 21-hydroxylase deficiency (21-OHD) is the corticotropin stimulation test (250 μg cosyntropin intravenously), measuring levels of 17-OHP at baseline and 60 minutes. These values can then be plotted on a nomogram to ascertain disease severity (Fig. 64-2). There is significant overlap between carriers and unaffected.
Assessment of fertility potential.
Electrolytes should be monitored closely for hyponatremia and hyperkalemia and treated immediately; salt-wasting is present in 75% of patients with classic 21-OHD and can be life threatening. Plasma renin and aldosterone ratio is elevated.
Extra doses of corticosteroids must be given during illness, trauma, and surgery to avoid crisis and death.
Hyperandrogenism is a clinical feature consistent in both 21-OHD and 11β-OHD.
46,XX females: Genital virilization occurs only in the androgen-responsive external genitalia. Since females with CAH have normal ovaries and do not produce anti-Müllerian hormone (AMH), the internal genitalia (uterus and fallopian tubes) develop normally from the Müllerian anlage. Genital virilization can vary greatly depending on the amount of androgens exposed to in utero. The degree of virilization is classified into five Prader stages:
Stage I: clitoromegaly without labial fusion
Stage II: clitoromegaly and posterior labial fusion
Stage III: greater degree of clitoromegaly, single perineal urogenital orifice, and almost complete labial fusion
Stage IV: increasingly phallic clitoris, urethra-like urogenital sinus at base of clitoris, and complete labial fusion
Stage V: penile clitoris, urethral meatus at tip of phallus, and scrotum-like labia (appear like males without palpable gonads)
Childhood: Depending on the amount of androgens exposed postnatally, an affected female may enter precocious pseudopuberty with early appearance of acne and axillary and pubic hair; rapid somatic growth with advanced epiphyseal maturation may lead to early epiphyseal closure and likely short stature.
Adults: Postpubertal females may develop progressive clitoral enlargement, deepening of the voice, increased muscle bulk, hirsutism, medication-resistant acne, and/or male pattern alopecia (temporal balding). Menstrual abnormalities, including oligomenorrhea or amenorrhea, and infertility may become evident.
46,XY males: In utero will not be affected by excess adrenal androgens and will thus be born with normal external genitalia which may be hyperpigmented.
Childhood: If left undetected males may also enter precocious pseudopuberty presenting similar to the females but with progressive penile enlargement but small volume of testes. Signs of excess adrenal androgens in the adult male may be difficult to assess, but may notably lead to decreased testicular size and testicular testosterone production as well as impaired spermatogenesis. These abnormalities are due to the effects of excess adrenal androgens on gonadotropin secretion.
Adult: A complication of inadequate hormonal control of CAH is hyperplastic nodular testes. Many adult male patients are found to have adenomatous adrenal rests within the testicular tissue. These tumors have been reported to be ACTH dependent and to regress following adequate steroid therapy. For males, these testicular adrenal rests that can also occur in females with salt-wasting CAH and are associated with an increased risk of infertility.
The degree of deficiency in 21-hydroxylase can be phenotypically categorized into two distinct forms: classic, which encompasses two clinical syndromes: salt-wasting and nonsalt-wasting (simple virilizer); and nonclassic. Females with nonclassic 21-OHD possess the biochemical defect but do not have ambiguous genitalia.
Salt-wasting 21-OHD: Along with the signs of hyperandrogenism, the newborn will present with failure to thrive, hyponatremia, hyperkalemia, inappropriate natriuresis, and high plasma renin activity due to low aldosterone within the first few weeks of life. If severe and untreated, hypotension, cardiac arrhythmias, vascular collapse, shock, and death can occur. As an adult these patients are fragile to illnesses, stressful events, and accidents. High doses of steroids are needed to prevent serious complications, such as shock and death.
Simple-virilizing 21-OHD: Comprises approximately 25% of the classic type. The neonate will produce enough aldosterone to prevent salt loss and its complications. If not picked up during newborn screening, affected males, who are born with normal genitalia, may present with precocious pseudopuberty from the effects of hyperandrogenism as described above.
Nonclassic 21-OHD: It is the most common form of CAH and can present at any age after birth. There is typically no genital abnormality in these females. Diagnosis may be made when evaluated for symptoms of excess androgens including early puberty or later in life in females for causes of infertility, menstrual abnormalities, hirsutism, obesity, cystic acne. Polycystic ovarian syndrome (PCOS) may also be seen as a secondary complication in these patients. Adult males may present with the signs of hyperandrogenism as described in the section earlier, but symptoms are often mild, and some patients may not present until being evaluated for infertility.
Along with the clinical presentation of excess androgen as described earlier, the key feature of 11β-OHD is suppressed renin and hypertension. Despite failure of aldosterone production, overproduction of the precursor deoxycorticosterone (DOC), a less potent in vivo mineralocorticoid, causes salt retention and hypertension. Elevated blood pressure is usually not identified until later in childhood or in adolescence and is loosely correlated with the elevation in DOC. The degree of virilization does not strongly correlate to blood pressure, and fatal vascular accidents can occur in minimally virilized patients. Maintaining good blood pressure control is important since complications of long-standing uncontrolled hypertension, including left ventricular hypertrophy, retinal vein occlusion, and blindness have been reported in 11β-OHD patients. Potassium depletion develops concomitantly with sodium retention, but hypokalemia is variable. Renin production is suppressed secondary to mineralocorticoid-induced sodium retention and volume expansion. Aldosterone production is low secondary to low serum potassium and low plasma renin. Diagnosis is made by genetic testing (DNA analysis).
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


