Key Points
Disease summary:
Pituitary tumors are typically of monoclonal origin (although not always); they are benign, often slow-growing, adenomas of the sella arising sporadically or rarely in the context of hereditary genetic syndromes accounting for 10% to 15% of all diagnosed intracranial neoplasms. Their true incidence is difficult to determine, as they are often asymptomatic. They can be effectively managed, as they are rarely malignant. Small pituitary tumors go largely undetected and are often only documented during postmortem studies; over 20% of the adult population may have a pituitary adenoma, identified incidentally by imaging studies (called “incidentalomas”). Significant morbidity occurs due to the pituitary tumor’s effect on hormone secretion and compression of regional structures. Other symptoms include those from mass effects.
Pituitary tumors can be classified as follows:
Common tumors of the sella turcica
Pituitary adenomas include nonfunctioning adenomas and tumors that hypersecrete hormones: growth hormone (GH)-omas (accounting for 20% of surgically treated lesions), prolactinomas (PRL; 50%), adrenocorticotropic hormone (ACTH)-producing adenomas (15%), and rarely thyroid-stimulating hormone (TSH)-omas, and gonadotropinomas (FSH-LH-omas).
Craniopharyngiomas are epithelial tumors arising from remnants of Rathke pouch and account for 3% of all intracranial tumors.
Supra- and parasellar meningiomas which account for 15% of all intracranial tumors.
Miscellaneous benign cysts: Rathke cleft cysts, intrasellar colloid cysts, arachnoid cysts.
Rare tumors of the sella turcica: Optico-hypothalamic gliomas, metastases, chordomas, inflammatory lesions, germinomas, hypothalamic harmartomas, chondromas, epidermoids.
Miscellaneous pituitary tumors: Granular cell tumor, paragangliomas, pituitary carcinomas (~0.2% operated pituitary neoplasms), mucocele, chiasmatic cavernoma, hypothalamic lipoma, and sarcoidosis.
Pathogenesis:
Pituitary tumors may be a manifestation of an underlying monogenic syndrome, such as McCune-Albright syndrome (MAS), multiple endocrine neoplasia type 1 (MEN1), Carney complex (CNC), multiple endocrine neoplasia type 4 (MEN4), and familial isolated pituitary adenomas (FIPA); alternatively, no single gene may be responsible. Some of these genes predispose to sporadic pituitary tumors, such as the aryl hydrocarbon receptor-interacting protein (AIP).
Twin studies:
Pituitary tumors are rarely inherited, however, in the very small number of these patients there is usually a family history of other endocrine (parathyroid or pancreas) tumors.
Environmental factors:
There are no known environmental causes.
Genome-wide associations:
A suggestive linkage on chromosome 19q13.41 has been identified as a possible modifier for the severity of acromegalic features in patients with isolated familial somatotropinoma.
Pharmacogenomics:
There are very few pharmacogenetic studies that assess the role of certain polymorphisms in the responsiveness to medications used in pituitary adenomas (eg, dopamine agonists and somatostatin analogues). NcoI T + genotype (homozygotes or heterozygotes for T allele), a D2 dopamine receptor (DRD2) polymorphism, has been associated with unresponsiveness to cabergoline treatment in patients affected with PRL-oma.
Diagnostic Criteria and Clinical Characteristics
The investigation of a patient presenting with evidence of a pituitary tumor has three main objectives:
Investigation of any hormonal hypersecretion
Assessment of residual pituitary function
Examination of any mass effect of the tumor (including headaches, visual disturbances, and cranial nerve palsies)
Diagnostic evaluation should include (Fig. 62-1 algorithm)
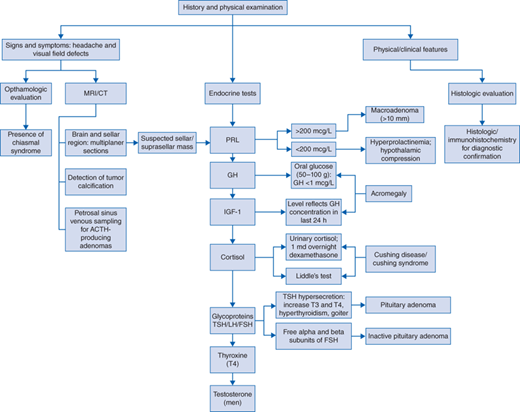
Visual fields and ophthalmologic evaluation in defining the presence of a chiasmal syndrome.
Initial screening endocrine tests should include levels of prolactin, insulin-like growth factor 1 (IGF-1), LH, FSH, TSH, and alpha subunit, cortisol, and thyroxine (T4); men should have their testosterone level checked.
Prolactin (PRL) levels
Serum PRL levels should be measured in any patient with a suspected sellar or suprasellar mass. If elevated, exclude the possibility of pharmacologic and other factors (eg, pregnancy) prior to requesting extensive neuroimaging studies.
Serum PRL level greater than 200 μg/L in a patient with an adenoma greater than 10 mm (macroadenoma) in size is diagnostic of a PRL-oma. Levels below this range in a macroadenoma suggest hyperprolactinemia secondary to hypothalamic compression.
GH levels
GH levels are elevated in acromegaly but can fluctuate significantly.
Serum IGF-1 level is the best endocrinologic test for acromegaly. IGF-1 reflects GH concentration in the last 24 hours.
Oral glucose tolerance test is the definitive test for the diagnosis of acromegaly; a positive result is the failure of GH to decrease to less than 1 μg/L after ingesting 50 to 100 g of glucose.
Cortisol levels—Cushing disease and Cushing syndrome
24-hour urinary free cortisol, 1 mg overnight dexamethasone suppression test, and midnight cortisol have all been recommended as the screening tests.
For the confirmation and differential diagnosis of Cushing disease, several other diagnostic tests have been developed including corticotropin-releasing hormone (CRH) stimulation test, the high-dose dexamethasone suppression test, and Liddle test.
Glycoprotein hormones—TSH, FSH, LH
Pituitary adenomas that are associated with TSH hypersecretion are uncommon. These patients have increased T3 and T4 levels, hyperthyroidism, and goiter with inappropriately high levels of TSH.
Free alpha and beta subunits of FSH are secreted by pituitary tumors thought to be inactive.
Magnetic resonance imaging (MRI) of the brain and sellar region with multiplanar thin sections is of critical importance. Pre- and postgadolinium images are recommended to ensure that primarily isointense lesions do not escape detection.
Computed tomographic (CT) scan of the brain with sellar images may be sufficiently specific and can detect tumor calcifications (particularly useful for the differential diagnosis of other parasellar masses, eg, craniopharyngioma).
Petrosal sinus venous sampling for ACTH-producing adenomas is performed in selective cases.
Standard histologic examination and immunohistochemistry of these lesions confirm the diagnosis. The mutated form of p53, a tumor suppressor, can also be determined histologically. The presence of the mutated p53 gene suggests a tumor with rapid, aggressive growth.
Pituitary microadenomas: Intrasellar adenomas less than 1 cm in diameter that present with manifestations of hormonal excess without sellar enlargement or extension. Panhypopituitarism does not occur and usually can be treated successfully.
Pituitary macroadenomas: Adenomas greater than 1 cm in diameter and cause sellar enlargement. Tumors 1 to 2 cm in diameter confined to the sella turcica can usually be treated successfully. Larger adenomas with suprasellar, sphenoid sinus, or lateral extension are more difficult to manage. Panhypopituitarism and visual loss are common findings.
GH-omas (also known as somatotropinomas)
Children and adolescents present with gigantism.
Adults presenting with acromegaly manifest (1) changes in the size of the hands and feet, coarseness of the face, frontal bossing, and prognathism; (2) macroglossia; (3) hypertrichosis, hyperpigmentation, and hyperhidrosis; (4) respiratory difficulty and sleep apnea; (5) hypertension and diabetes mellitus; (6) cardiac complications resulting from acromegalic cardiomyopathy; (7) carpal tunnel syndrome; (8) colonic polyps (association with colon cancer).
PRL-omas
Women: amenorrhea, galactorrhea, and infertility
Men: decreased libido, impotence, rarely galactorrhea
ACTH-producing adenomas (Cushing disease)
Weight gain, centripetal obesity, “moon face,” violet striae, easy bruisability, proximal myopathy, and psychiatric changes
Other possible effects including arterial hypertension, diabetes, cataracts, glaucoma, and osteoporosis
TSH-omas
Hyperthyroidism with goiter (enlarged thyroid gland) in the presence of elevated TSH levels, the presence of headache as well as visual field defects are attributed to tumor expansion.
Gonadotropinomas (FSH-LH-omas)
Usually large chromophobe adenomas presenting with visual impairment due to tumor mass effect. Rarely, these tumors are associated with precocious puberty or resumption of bleeding in postmenopausal women.
Nonfunctioning adenomas
Headache and visual field disturbances are the usual presenting symptoms.
Endocrine manifestations often precede the diagnosis for months or years, but the symptoms are subtle and maybe missed. The most common are hypogonadism, hypothyroidism, and hypoadrenalism.
MEN1
Because of the low prevalence of the syndrome, MEN1 is responsible for only 2.7% of all pituitary adenomas.
About 40% of patients with MEN1 have pituitary adenoma.
Pituitary tumors in MEN1 appear to be larger and more aggressive than in patients without MEN1, with macroadenomas being present in 85% of the former, compared with only 42% of the sporadic cases.
PRL-omas predominate among both MEN1-associated adenomas and non-MEN1 pituitary adenomas. MEN1-related PRL-omas are usually macroadenomas (84%), and show higher rates of invasion than in non-MEN1 PRL-omas.
MEN1-associated pituitary tumors are significantly more likely to cause symptoms due to tumor size and have a significantly lower rate of hormonal normalization than non-MEN1.
MAS arises from an activating gsp mutation in the GNAS1 gene located on chromosome 20q13.2.
Pituitary lesions are usually hyperplastic and are less commonly adenomatous.
GH hypersecretion in MAS differs from the classical acromegaly; patients are generally young at the onset of the disease (<20 years) and diagnosis is usually based on growth acceleration rather than dysmorphic features, which are difficult to assess owing to fibrous dysplasia present in the syndrome.
FIPA
This familial condition is characterized by pituitary tumors of all types in members of a single kindred in the absence of mutations in genes related to MEN1 or CNC.
In FIPA, tumors may be of homogeneous phenotype within a family or members of the same family may have different pituitary tumor types (heterogenous).
Pituitary tumors in patients with FIPA tend to present 4 years earlier than the sporadic ones, are significantly larger, and tend to have a higher rate of invasion in the cavernous sinus.
Mutations of the AIP gene (aryl hydrocarbon receptor-interacting protein) may account for 15% of FIPA families.
CNC
The pituitary is one of multiple endocrine organs affected in CNC, and features hypersomatotropinemia and hyperprolactinemia, often beginning in adolescence. Clinically evident acromegaly due to a GH-producing tumor is a relatively infrequent manifestation of CNC. As with MAS, somatomammotrophic hyperplasia, a precursor of GH-producing adenoma, may precede clinically evident acromegaly in CNC patients.
MEN4
Two families have been reported harboring a mutation in the CDKN1B gene:
A German family exhibiting acromegaly, primary hyperparathyroidism, renal angiomyolipoma, and testicular cancer among various members
A Dutch patient with a pituitary adenoma (Cushing disease), a cervical carcinoid tumor, and hyperparathyroidism
Screening and Counseling
Pituitary tumors are of endocrine origin and occur sporadically and for the most part remain unknown. The hereditary risk to relatives is not generally increased should a single family member present with a particular tumor (Fig. 62-1).
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


