Key Points
Disease summary:
Multiple endocrine neoplasia type 2 (MEN2) is caused by a dominantly inherited or de novo activating (gain of function) mutation in the RET proto-oncogene.
MEN2A (95% of MEN2 cases) is characterized by the development of medullary thyroid carcinoma (MTC) in greater than 90% of affected patients, pheochromocytoma (PHEO) in up to 50% of cases, and/or primary hyperparathyroidism (PHPT) in up to 20% of mutation carriers. Depending on the specific RET mutation, cutaneous lichen amyloidosis (CLA) and Hirschsprung disease can also occur.
MEN2B (5% of MEN2 cases) is characterized by the universal and early development of MTC, high risk of PHEO (up to 50% of cases) and a highly penetrant, distinctive physical appearance.
A strong genotype-phenotype correlation exists in MEN2 such that MTC disease severity, the likelihood of developing PHEO and PHPT, and the age of disease onset can be estimated based on genetic testing results.
In RET mutation carriers, C-cell hyperplasia is the initial stage of tumor development that leads to microscopic noninvasive MTC (usually bilateral) and ultimately to lymph node and distant metastases due to frankly invasive carcinoma.
Familial MTC (FMTC) is currently considered to be a phenotypic variant of MEN2A with a high risk for MTC but decreased penetrance and/or delayed onset of the other neoplastic manifestations. There is significant overlap between RET mutations associated with FMTC and those of MEN2A.
Hereditary basis:
MEN2A and MEN2B have an autosomal dominant inheritance pattern with almost complete penetrance of the MTC phenotype.
Differential diagnosis:
There is no other genetic syndrome associated with the development of MTC. Other multiorgan system syndromes that include PHEO as a feature include von Hippel-Lindau disease, the familial paraganglioma syndromes, and neurofibromatosis type 1. Familial PHPT is most commonly associated with MEN1 but the differential diagnosis also includes familial isolated primary hyperparathyroidism, familial hypocalciuric hypercalcemia, and hyperparathyroidism-jaw tumor syndrome. Hirschsprung disease can be sporadic or associated with underlying chromosomal abnormalities and gene mutations, including inactivating (loss of function) RET mutations that are distinct from the MEN2-causing mutations. The differential diagnosis for the skeletal phenotype of MEN2B includes Marfan syndrome and homocystinuria; intestinal ganglioneuromatosis may also be associated with Cowden syndrome and type 1 neurofibromatosis.
Diagnostic Criteria and Clinical Characteristics
At least one of the following
One or more of the MEN2-associated endocrine tumors in a patient who has an identified germline mutation in the RET proto-oncogene
Identification of a known MEN2-causing germline mutation in the RET proto-oncogene, particularly with a positive family history of any of the known MEN2-related endocrine tumors
A diagnosis of MTC in a patient who has one or more close relatives with histologically confirmed MTC
A clinical diagnosis of MEN2 (prior to confirmation via genetic testing) would be highly suspected in a given patient with two or more of the endocrine tumors associated with MEN2 or a diagnosis of MTC in a patient with the clinical features of MEN2B
And the absence of
Another identifiable multiorgan system genetic syndrome that explains the development of PHPT, PHEO, and/or physical examination findings in the proband or family
(Table 59-1 and Table 59-2)
| System | Manifestation | Comments |
|---|---|---|
| Thyroid C cells | Medullary thyroid carcinoma | >90% of MEN2A cases |
| Adrenal medulla | Pheochromocytoma | 0%-50% of MEN2A cases |
| Parathyroid glands | Primary hyperparathyroidism | 0%-20% of MEN2A cases |
| Skin | Cutaneous lichen amyloidosis | Rare |
| GI tract | Hirschsprung disease | Rare |
| System | Manifestation | Comments |
|---|---|---|
| Thyroid C cells | Medullary thyroid carcinoma | 100% of MEN2B cases |
| Adrenal medulla | Pheochromocytoma | ~50% of MEN2B cases |
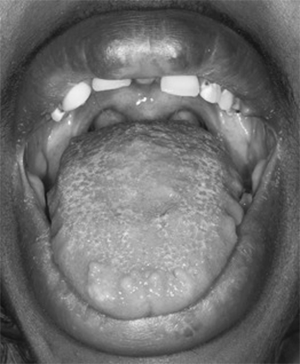
| GI tract | Oral mucosal (Fig. 59-1) and intestinal ganglioneuromatosisa | ~100% of MEN2B cases |
| Skeleton | Marfanoid body habitus, narrow long facies, pescavus, pectus excavatum, high-arched palate, scoliosis, and/or slipped capital femoral epiphysis | ~75% of MEN2B cases |
| Eyes | Inability to make tears in infancy, thickened and everted eyelids, mild ptosis, and prominent corneal nerves (medullated corneal nerve fibers) | Varies |
| Other | Joint laxity; hypotonia or proximal muscle weakness, thickened lips; pubertal delay | Varies |
Arising from the parafollicular C cells, MTC is usually the first MEN2 manifestation. Years before clinical disease becomes apparent, MTC begins as C-cell hyperplasia, a precancerous finding that ultimately develops into microscopic and then macroscopic cancer that is typically multifocal, bilateral, and located in the middle to upper regions of the thyroid lobes. Clinical presentations can include a palpable thyroid and/or neck mass, compressive symptoms, or diarrhea related to elevated calcitonin levels and other substances secreted by the tumor. Some patients may have microscopic MTC identified incidentally during thyroid surgery for another indication and, with the onset of genetic testing, more individuals are being identified to have lesser degrees of disease due to early surgical intervention at a presymptomatic stage. Lymph node and distant metastases typically occur years after the onset of tumorigenesis, and the most aggressive clinical presentation occurs with MEN2B, followed by codon 634 mutations in MEN2A, and finally by the other known disease-causing mutations in RET. Cervical and mediastinal lymph nodes are the most common sites of metastatic disease; typical distant sites for MTC spread include the lungs, liver, and bone or bone marrow. Calcitonin and carcinoembryonic antigen (CEA) are excellent tumor markers for MTC, with rapid doubling times associated with more aggressive disease and a worse prognosis.
The classic triad of clinical features includes diaphoresis, headache, and palpitations in a patient with episodic or sustained hypertension, but patients with PHEO may not have these classic features and may even be entirely asymptomatic. PHEO in patients with MEN2 usually arise within a background of adrenal medullary hyperplasia and are associated with an adrenergic clinical phenotype (elevation of epinephrine ± norepinephrine, in addition to their metabolites, metanephrine and normetanephrine, respectively). Individuals with MEN2 are at an increased risk for bilateral PHEO and an earlier age of onset, and these tumors may present synchronously or even prior to an MTC diagnosis, especially in MEN2A. Paragangliomas (catecholamine-producing tumors located outside of the adrenal gland) and malignant tumors are exceedingly rare in MEN2.
PHPT in patients with MEN2A may be due either to a single parathyroid adenoma or multigland hyperplasia. The disease is diagnosed and evaluated similarly to sporadic PHPT.
CLA is skin disorder of intense pruritus and secondary skin changes typically located in interscapular region of the back that becomes clinically manifest in some MEN2A patients during late adolescence or young adult life.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


