Key Points
Disease summary:
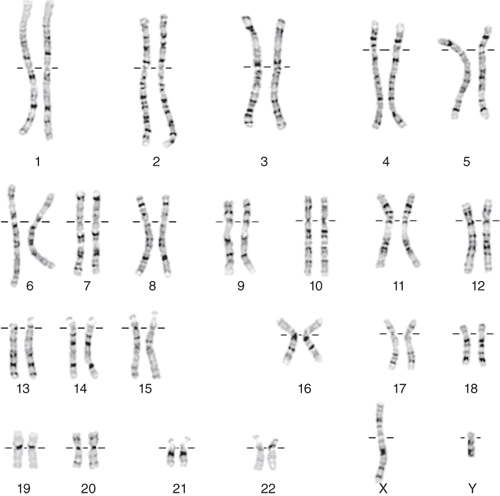
Cytogenetics is the study of chromosome structure, function, and disorders by combining cytology (the study of cells) with clinical genetics (the study of inherited variation). The normal diploid complement of chromosomes consists of 46 chromosomes: 22 pairs of numbered autosomal chromosomes and one pair of lettered sex chromosomes (X and Y) (Fig. 167-1). One haploid complement is composed of 23 chromosomes, inherited from the mother in the egg or the father in the sperm. A complete diploid set of 46 chromosomes is present in the zygote, and upon replication, they are present in every cell nucleus of the offspring. A normal chromosome complement (or karyotype) is designated as 46,XX for females and 46,XY for males. Chromosomes each have a short arm (p) and a long arm (q). Chromosome regions as well as gene locations are annotated as the chromosome number followed by “p” or “q” (for the short or long arm, respectively) followed by the band number (organized by the banding pattern chromosomes uniformly display on in vitro staining). During metaphase the region between the p and q arms is condensed and constricted and is designated the centromeric region where the spindle apparatus is attached. Some chromosomes such as chromosome 1, 6, 7, 11, 14, 15, 18, 19, and 20 contain regions that are differentially methylated, or “imprinted,” based on the parent of origin; imprinted portions of chromosomes are transciptionally silenced, meaning genes in those segments are not translated to protein. The term chromosome disorder refers to a clinical condition secondary to an abnormality in the quantity, content, and/or arrangement of the 23 pairs of chromosomes comprised of the nuclear genomic DNA.
Chromosomal abnormalities (see Table 167-1):
Aneuploidy, alteration in the total number of chromosomes, such as trisomy and monosomy
Euploidy, a multiple of the normal number of chromosomes, 46 (eg, 69,XXX or 92,XXXX, which are embryonic lethal)
Deletion, loss of a portion of a chromosome (eg, DiGeorge or velocardiofacial syndrome due to deletion of 22q11.2, or chromosome 22, q arm region, band 11.2 [designated “one one point two”])
Duplication, gain of a portion of a chromosome (eg, Charcot-Marie-Tooth type 1 due to duplication of the PMP22 gene in chromosome17p12)
Insertion, gain of a portion of a chromosome
Pericentric inversion, inversion of a portion of a chromosome including the centromeric region
Paracentric inversion, inversion of a portion of a chromosome arm not including the centromeric region
Robertsonian translocation, fusion of two acrocentric chromosomes 13, 14, 15, 21, or 22 (chromosomes with a very small p arm) with breakpoints at or near the centromere with loss of repeated DNA sequences from the short p arms that usually is without clinical consequence. A karyotype with a robertsonian translocation has a total of 45 chromosomes.
Reciprocal translocation, rearrangement of portions of two different chromosomes formed by the reciprocal exchange between portions from autosomal or sex chromosomes (Fig. 167-2)
Isochromosome, abnormal arrangement of chromosome arms with either fusion of two p arms separated by a centromere without a q arm or fusion of two q arms separated by a centromere without a p arm forming a chromosome
Ring chromosome, abnormal arrangement of a chromosome with breakage and fusion in the p arm and q arm to form a ring
Uniparental disomy, inheritance of both maternal and paternal copies of the same chromosome, instead of the normal inheritance of one maternal and one paternal chromosome forming a complementary pair. This may result in an abnormal phenotype or a particular syndrome if the chromosome involved is imprinted (eg, Prader-Willi syndrome due to maternal uniparental disomy of chromosome 15)
Trinucleotide repeat expansion, a sequence of three nucleotides that may not cause symptoms in the “premutation or carrier” status, but may expand in number of repeats during gametogenesis and lead to disease in an offspring
Other variant chromosomes, telomeric fusions and complex rearrangements
Chromosome disorders
These are either present from embryogenesis and are constitutionally present in the individual (germline) or develop over time as a result of DNA replication errors and dysfunctional DNA repair (somatic); the latter are important in cancer biology.
Constitutional chromosome abnormalities that may not be detected until adulthood are usually rearrangements in which no significant DNA material is lost or gained. These alterations are termed balanced, and usually do not result in phenotypic abnormality unless a gene is interrupted in the breakpoints of the rearrangement, or regulatory elements for a gene are disturbed (positional effect).
Cancer chromosomal abnormalities include hundreds of different mutations, many of which are recurrent aneuploidies and reciprocal translocations. Chromosome mutations that accumulate in cancer are acquired, somatic genetic changes that generally interrupt or activate oncogenic or tumor suppressor loci.
Certain translocations carry prognostic import for responsiveness to therapy, rate of recurrences, and survival, particularly in hematologic malignancies.
Certain chromosomes have fragile sites prone to an abnormal thin or stretched appearance when grown in folate-deficient media that may be correlated with clinical syndromes depending on the chromosome location. Common fragile sites that are inherited or of unknown clinical significance can be induced with aphidicolin or 5-azacytidine.
Chromosome breakage syndromes demonstrate chromosome fragility and structural abnormalities on chromosome analysis and are often diagnosed in childhood due to heightened cancer disposition and immune malfunction.
Mosaicism refers to the existence of more than one distinct chromosomal constitution within cells of one or multiple tissue types.
All females are mosaic with two different cell types because one X chromosome (maternally or paternally derived) is actively transcribed and the other X is randomly inactivated (or lyonized) forming a Barr body. Inactivation is propagated along the inactive X chromosome by the expression of Xist RNA from the inactive X, followed by chromatin condensation and methylation.
A chromosomal abnormality may be found as a tissue-limited mosaic line and result in mild or no features of the full chromosomal disorder (eg, trisomy 21 may be found in certain tissues and not others, resulting in a mild or no features of Down syndrome; or mosaicism for X chromosome rearrangements with interruption of genes in particular chromosome locations, or loci, resulting in the neurocutaneous syndrome, hypomelanosis of Ito).
Technology to analyze chromosomes has evolved from gross examination of metaphase chromosomes (karyotype) at a resolution from about 5 Mb (million DNA base pairs) to a submicroscopic resolution of about 50 to 200 kb using fluorescently labeled probes for specific chromosome regions, fluorescent in situ hybridization (FISH), and whole genome dosage examination at a resolution of at least 10 kb using array comparative genomic hybridization (array CGH, or microarray). Array resolution depends on the density of probes (either single-nucleotide polymorphisms, SNPs, or stretches of DNA, known as oligonucleoties) tiled along the microarray chip.
Microdeletions and microduplications comprise a set of well-characterized syndromes due to deletion or duplication of a chromosomal segment or region and thereby haploinsufficiency or triplosufficiency for the dosage-sensitive genes in that locus. Many are recurrent at a predictable rate in the population, and are caused by aberrant recombination between repeated sequences or nonallelic homologous recombination.
Copy number variants (CNVs) are the most common type of structural variation in the human genome, representing regions of chromosomes that are deleted or duplicated. Some CNVs are de novo (arised during gametogenesis or embryogenesis and present newly in offspring) and may be associated with clinical significance, whereas others may be inherited usually with no phenotypic consequences.
Medical history:
Chromosomal disorders that come to clinical attention in adults are often detected during workup for pubertal delay, reproductive problems, abnormalities in an offspring, or cancer
Primary amenorrhea in a female
Oligospermic or nonobstructive azoospermic infertility (failure to conceive regardless of age after 1 year of unprotected intercourse) in a male
Infertility in a female (failure to conceive regardless of age after 1 year of unprotected intercourse)
Recurrent miscarriage (three or more consecutive spontaneous abortions)
Chromosomal disorder detected in a child (usually on workup for multiple congenital anomalies and developmental delay)
Balanced translocation (with no DNA lost or gained) carriers are at risk to pass on an unbalanced derivative chromosome leading to an abnormal phenotype in an offspring inheriting what appears as grossly the same translocation (imbalance detected on karyotype or higher-resolution analysis, such as array CGH)
Hematologic malignancy, Wilms tumor
Family history:
Recurrent miscarriage
Two or more first- (children, siblings, parents) or second-degree relatives (grandparents, aunts, uncles) with multiple congenital anomalies and mental retardation
Multiple cases of cancer at young ages of diagnosis and/or immunologic defects
Differential diagnosis:
Other causes of delayed puberty include endocrine disorders.
Other causes of female infertility include endocrine disorders, gynecologic factors, metabolic diseases, infectious etiologies.
Other causes of male infertility include cystic fibrosis mutation.
Pharmacogenomics:
Use of imatinib mesylate (Gleevec) for chronic myelogenous leukemia is based on the presence of the Philadelphia chromosome translocation (between chromosomes 9 and 22) and a hyperactive bcr-abl protein. The drug inhibits the chimeric protein in cancerous cells.