Key Points
Disease summary:
Hearing loss (HL) is the most common sensory deficit in humans, occurring in 1 in 500 births and affecting 278 million people worldwide.
Conductive HL (CHL) is characterized by abnormalities of the external and/or middle ear, while sensorineural hearing loss (SNHL) is caused by malfunction of the inner ear, and mixed HL is a combination of both CHL and SNHL.
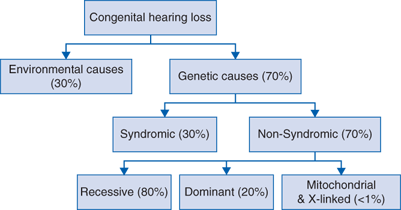
Nonsyndromic HL (NSHL) is not associated with any abnormalities of the external ear or any other organs and comprises the majority (70%) of hereditary HL (Fig. 151-1).
Hereditary basis:
Most cases of NSHL have a genetic etiology.
NSHL is genetically heterogeneous with 64 genes currently implicated in the disorder (Table 151-1).
Mutations in the gene encoding the gap junction protein connexin 26 (GJB2) are the most common genetic cause of NSHL, occurring in an estimated 25% of autosomal recessive cases in the US population.
Differential diagnosis:
Environmental causes of hearing impairment (acquired HL) account for approximately 30% of NSHL in developed countries
Acquired HL in children may be caused by
Prenatal infections from TORCH organisms (Toxoplasmosis, Rubella, Cytomegalovirus [CMV], and Herpes) with infection comprising the majority of environmental HL in children
Postnatal infections, particularly bacterial meningitis caused by Neisseria meningitides, Haemophilus influenzae, or Streptococcus pneumoniae
Acquired HL in adults is most often due to environmental noise exposure, though there is evidence for gene-environment interactions in “age-related” HL.
Syndromic hearing impairment is characterized by hearing loss and abnormal findings of at least one other organ system. There are more than 400 genetic syndromes that include hearing impairment and only the most common are briefly outlined here.
Autosomal dominant syndromic hearing impairment
Waardenburg syndrome: variable SNHL, pigmentary abnormalities including white forelock, and heterochromia iridis
Branchiootorenal syndrome (BOR syndrome): CHL, SNHL, or mixed HL, branchial cleft cysts, malformations of the external ear, and renal anomalies
Stickler syndrome: progressive SNHL, cleft palate, and spondyloepiphyseal dysplasia leading to arthritis
Autosomal recessive syndromic hearing impairment
Usher syndrome (USH): the most common cause of deaf-blindness is characterized by congenital SNHL, retinitis pigmentosa, and vestibular dysfunction. There are three types of USH differentiated by hearing and vestibular phenotypes:
USH type I: severe-to-profound SNHL, vestibular deficits
USH type II: mild-to-severe SNHL, normal vestibular function
USH type III: progressive SNHL and progressive loss of vestibular function
Pendred syndrome: severe-to-profound SNHL and euthyroid goiter associated with an abnormality of the bony labyrinth on CT examination (Mondini dysplasia or enlarged vestibular aqueduct [EVA])
Jervell and Lange-Nielsen syndrome: congenital deafness and prolongation of the QT interval leading to syncopal episodes
X-linked syndromic impairment
Alport syndrome: progressive SNHL and progressive glomerulonephritis which can lead to end-stage renal disease along with variable ophthalmologic findings (eg, anterior lenticonus)
NSHL is distinguished by a lack of visible abnormalities of the external ear or other organ systems, however, it can be associated with abnormalities of the middle and/or inner ear (Table 151-1). NSHL is categorized by mode of inheritance and the different gene loci are designated DFN (for DeaFNess) and then given a letter according to inheritance pattern.
Autosomal dominant NSHL (DFNA): generally progressive and moderate to severe. Significant genotype-phenotype correlations exist (eg, WFS1, DFNA6/14, causes low-frequency SNHL).
Autosomal recessive NSHL (DFNB): generally congenital severe to profound, with exceptions (eg, TECTA, DFNB21, causes moderate NSHL).
X-linked NSHL (DFNX): there are two identified X-linked NSHL genes with variable phenotypes.
Mitochondrial NSHL: mutations in two mitochondrial genes cause NSHL by a mechanism that is not understood and some cases (eg, MT-RNR1 mutations) susceptibility to developing deafness is conferred by aminoglycoside administration.
| Gene | Deafness Locus | Full Name | OMIM # | Phenotype |
|---|---|---|---|---|
Autosomal Recessive NSHL Genesa | ||||
BSND | DFNB73 | Barttin | 606412 | Prelingual, severe |
CDH23 | DFNB12 | Cadherin-related 23 | 601386 | Prelingual, relatively stable |
CLDN14 | DFNB29 | Claudin 14 | 605608 | Prelingual, profound |
COL11A2 | DFNB53/DFNA13 | Collagen, type XI, alpha 2 | 609706 | Postlingual (second decade), mid frequency |
ESPN | DFNB36 | Espin | 609006 | Prelingual, vestibular areflexia |
ESSRB | DFNB35 | Estrogen-related receptor beta | 608565 | Prelingual, profound |
GIPC3 | DFNB15/DFNB95 | GAIP C-terminus interacting protein 3 | 601869 | Prelingual, mid-high frequency |
GJB2 | DFNB1/DFNA3 | Gap junction protein, beta 2 | 220290 | Prelingual, relatively stable, all frequencies or high frequency |
GJB3 | DFNA2 | Gap junction protein, beta 3 | 600101 | Details not available |
GJB6 | DFNB1/DFNA3 | Gap junction protein, beta 6 | 220290 | Details not available |
GPSM2 | DFNB82 | G-protein signaling modulator 2 | 613557 | Prelingual, stable, profound |
GRXCR1 | DFNB25 | Glutaredoxin cysteine-rich 1 | 613285 | Prelingual, severe to profound |
HGF | DFNB39 | Hepatocyte growth factor | 608265 | Prelingual, severe to profound |
ILDR1 | DFNB42 | Ig-like domain-containing receptor 1 | 609646 | Prelingual, mid-to-high frequency |
LHFPL5 | DFNB66/67 | Lipoma HMGIC fusion partner-like 5 | 610212 | Prelingual, severe to profound |
LOXHD1 | DFNB77 | Lipoxygenase homology domain-containing 1 | 613079 | Prelingual, mid-to-high frequency, progressive |
LRTOMT | DFNB63 | Leucine-rich transmembrane OMT | 611451 | Prelingual, profound |
MARVELD2 | DFNB49 | MARVEL domain-containing 2 | 610153 | Prelingual, moderate to profound |
MSRB3 | DFNB74 | Methionine sulfoxide reductase B3 | 613719 | Prelingual, profound |
MYO3A | DFNB30 | Myosin III A | 607101 | Progressive, affects high then mid frequencies |
MYO6 | DFNB37/DFNA22 | Myosin VI | 607821 | Prelingual or postlingual high frequency |
MYO7A | DFNB2/DFNA11 | Myosin VII A | 600060 | Prelingual or postlingual (first decade) |
MYO15A | DFNB3 | Myosin XVA | 600316 | Prelingual, stable, severe to profound |
OTOA | DFNB22 | Otoancorin | 607039 | Prelingual, moderate to severe |
OTOF | DFNB9 | Otoferlin | 601071 | Prelingual, stable |
PCDH15 | DFNB23 | Protocadherin-related 15 | 609533 | Prelingual, severe to profound |
PJVK | DFNB59 | Pejvakin | 610220 |
|



