Key Points
Disease summary:
Primary open angle glaucoma (POAG) is a genetically and clinically complex disease with multiple genetic risk factors and environmental exposure influencing disease susceptibility.
POAG is defined by progressive degeneration of the optic nerve. Elevation of intraocular pressure (IOP) is a risk factor for optic nerve degeneration, however, approximately 30% of POAG patients have optic nerve degeneration despite IOPs in the normal range (called normal-pressure glaucoma or NPG, also normal-tension glaucoma or NTG).
Ocular traits other than IOP also contribute to POAG risk including the thickness of the central cornea, the size of the optic nerve (optic nerve area), and the size of the optic nerve “cup” relative to the overall size of the nerve (optic nerve cup-to-disc-ratio, CDR).
Differential diagnosis:
Exfoliation glaucoma, pigment dispersion glaucoma, developmental glaucoma, angle-closure glaucoma
Monogenic forms:
Autosomal dominant juvenile-onset primary open-angle glaucoma caused by mutations in the MYOC gene.
Family history:
Siblings and first-degree relatives have a 7 to 10 times increased risk of developing POAG overall.
Twin studies:
Monozygotic twins have an increased risk of disease when compared to dizygotic twins.
Environmental factors:
Postmenopausal hormone use and body mass index (BMI) have been suggested as environmental risk factors for POAG.
Genome-wide associations:
Genome-wide association studies (GWAS) have identified five genes or loci for POAG: CDKN2BAS (POAG and NPG), CAV1/CAV2 (POAG), TMCO1 (POAG), SIX1/SIX6 (POAG), and 8q22 (NPG). GWAS for ocular quantitative traits relevant to POAG have also yielded results: CDKN2BAS (CDR), SIX1/SIX6 (CDR), ATOH7 (optic nerve area), and a number of genes or loci for central corneal thickness (CCT), a trait that is a risk factor for the disease. Disease-associated genetic variants (single-nucleotide polymorphisms [SNPs]) have provided insight into disease pathogenesis; testing for SNPs is not yet clinically validated to diagnose or guide management of POAG.
Pharmacogenomics:
Testing for common variants has not yet been shown to influence management of POAG.
Diagnostic Criteria and Clinical Characteristics
Diagnostic evaluation should include the following (Fig. 148-1 algorithm):
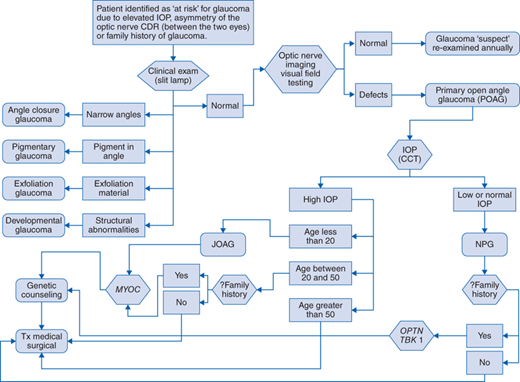
Clinical examination of the ocular anterior segment (cornea, trabecular meshwork, iris, lens) using the slit lamp
Evaluation of the optic nerve by fundoscopy and other imaging modalities (optic nerve photography, laser scanning imaging [HRT], ocular computed tomography [OCT])
Visual field evaluation
Measurement of IOP
Measurement of CCT
And the absence of
Narrow angles (angle-closure glaucoma)
Exfoliation material (pseudoexfoliation syndrome)
Increased pigment in the trabecular meshwork (pigment dispersion syndrome)
Absence of transillumination defects in the iris (pigment dispersion syndrome)
Developmental abnormalities of the anterior segment (developmental glaucoma)
A patient potentially at risk for glaucoma is identified through general eye care screening because of elevated IOP (>21 mm Hg), asymmetry of the optic nerve CDR, or a family history of disease. The patient undergoes further clinical examination of the ocular anterior segment (slit-lamp examination). For some types of glaucoma, specific anatomic abnormalities will establish a diagnosis other than POAG. If the ocular anterior segment is anatomically normal then the patient undergoes optic nerve imaging and visual field testing. If detects are not detected the patient is considered a glaucoma suspect and is re-evaluated on an annual basis. If visual field and/or optic nerve defects are identified the patient is given a diagnosis of POAG. After IOP and CCT testing, if the patient has high IOP and disease onset was at 20 years old or younger they are given the diagnosis of juvenile open-angle glaucoma (JOAG) and are referred for MYOC gene testing. If the patient has high IOP and is between 20 and 50 years of age at disease onset and also has a family history of disease, referral for MYOC gene testing is indicated. All patients with MYOC mutations and/or a family history of glaucoma are referred for genetic counseling. Patients older than 50 years of age at time of disease onset are less likely to have MYOC mutations. All patients with POAG are treated with medications and possibly surgery to lower the IOP. For POAG patients with low IOP (≤21 mm Hg), they are given the diagnosis of NPG and if there is a family history of NPG they are referred for OPTN and TBK1 gene testing. All patients with gene mutations and/or a family history are referred for genetic counseling. All patients with NPG are treated with medications and possibly surgery to lower IOP.
Glaucoma causes progressive painless degeneration of the optic nerve. If left untreated, the optic nerve damage results in irreversible and complete blindness. The disease onset is typically in the sixth and seventh decades, although individuals may be affected by age 40 and patients carrying mutations in MYOC develop an early-onset form of the disease during the first and second decades of life. Loss of vision typically begins in the peripheral visual fields where it may not be noticed by the patient. Gradually the peripheral visual field defects progress to involve the high acuity central visual fields. In POAG, elevation of IOP precedes the damage to the optic nerve and IOP screening can identify individuals at early disease stages when treatment is most effective. IOP measurement should be accompanied by measurement of the CCT because the corneal thickness can influence the IOP measurement, and studies suggest that low CCT (<530 nm) is an independent risk factor for POAG. High IOP is typically greater than 21 mm Hg while “normal” IOP is less than or equal to 21 mm Hg. In the NPG form of POAG, without elevated IOP, patients may initially present for treatment at advanced stages of the disease with severe optic nerve damage.
Patients that develop POAG as children (juvenile-onset POAG also known as JOAG with onset between the ages of 3 and 20) typically have very high IOP (>25 mm Hg) and rapidly progressive optic nerve disease. Children who are carriers of MYOC mutations frequently require surgery to control the IOP.
Screening and Counseling
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


