Key Points
Disease summary:
Alzheimer disease (AD) is the most common form of dementia. It is characterized by debilitating and progressive episodic memory loss, difficulty with language and decision making, and (in advanced stages) loss of motor control, incontinence, and mutism.
The neuropathologic hallmarks of the AD brain include an abundance of senile plaques largely composed of beta-amyloid (Aβ) deposits and neurofibrillary tangles made up of tau protein. While these features are consistently observed in autopsies of AD patients, the exact role that these proteins play in AD pathogenesis is still unclear.
Most patients with AD begin developing symptoms after 60 years of age, although in the rare cases of autosomal dominantly inherited AD symptoms typically manifest at an earlier age.
Although risk for AD increases with age, AD is not a symptom of normal aging.
Differential diagnosis:
Treatable diagnoses: depression, chronic drug intoxication, chronic central nervous system (CNS) infection, thyroid disease, vitamin deficiencies (particularly B12 and thiamine), CNS angiitis, and normal-pressure hydrocephalus (NPH)
Other neurodegenerative disorders: vascular dementia, diffuse Lewy body syndrome, Parkinson disease, Pick disease, Creutzfeldt-Jakob disease, and cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy (CADASIL)
Monogenic forms:
Rare cases of very early-onset familial AD (FAD) follow a Mendelian autosomal dominant inheritance pattern.
Approximately 70% of FAD cases can be explained by mutations in one of three genes: PSEN1, PSEN2, and APP.
Family history:
The average person has a 10% to 12% chance of developing AD in his or her lifetime. First-degree relatives of patients with AD have a 20% to 44% lifetime risk for the disease.
Twin studies:
Monozygotic twins show a 60% concordance rate for AD.
Environmental factors:
Risk factors include female gender, lower level of education, and history of head trauma.
Genome-wide Associations:
Recent large-scale genome-wide association studies (GWASs) have identified multiple genetic loci associated with AD risk (Table 124-1). Of these results however, even the most robust marker for AD susceptibility, the ε4 allele of APOE, is neither necessary nor sufficient for AD diagnosis.
Pharmacogenomics:
While several studies have begun stratifying AD patients based on genetic markers to test for differential response to treatment, at present no consistently replicated pharmacogenomic associations have been observed for AD therapies.
| Gene (Location) | Name | Form of AD | Inheritance Pattern | Penetrance | Available Testing |
|---|---|---|---|---|---|
| APPa(21q21.3) | Amyloid precursor protein | FAD | Autosomal dominant | 100% | Clinically available for adults but rare |
| PSEN1(14q24.2) | Presenilin 1 | FAD | Autosomal dominant | 100% | Clinically available for adults and prenatal testing |
| PSEN2(1q42.13) | Presenilin 2 | FAD | Autosomal dominant | 95% | Clinically available for adults but rare |
| APOE(19q13.32) | Apolipoprotein E | Sporadic AD | Complex | N/A | Clinically available for adults through direct-to-consumer testing |
Diagnostic Criteria and Clinical Characteristics
Making a diagnosis of AD can be particularly challenging (see Fig. 124-1), since a definitive diagnosis of AD requires autopsy results demonstrating a large number of Aβ neuritic plaques and neurofibrillary (tau protein) tangles in the brain. However, clinical criteria for making the diagnosis were established in 1984 and have proven highly accurate (81% sensitivity, 70% specificity). To meet the clinical criteria, the patient must have gradual progressive cognitive decline (as opposed to acute onset of symptoms) and cognitive impairment must be observed in at least two of the following domains:
Impaired ability to learn or recall information
Impaired language skills
Impaired visuospatial abilities
Executive dysfunction
Changes in behavior or personality
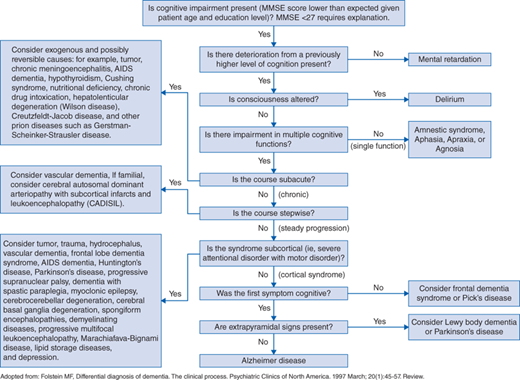
More recent criteria for the clinical diagnosis of AD have described AD as part of a continuum of biologic phenomena starting from a preclinical phase then progressing to a phase of mild cognitive impairment and later dementia. Supportive evidence of this model has been demonstrated from a longitudinal study of families with a history of autosomal dominant AD, which showed that physiologic changes in affected brains are observable up to 20 years prior to the onset of dementia and 10 years before the earliest cognitive symptoms manifest. These data suggest that previous diagnoses of AD were made late in the course of disease progression and provide a strong case for earlier diagnosis by applying several methods of biomarker detection discussed later in addition to standard clinical assessments for monitoring cognitive decline.
AD typically presents in patients between 40 and 90 years of age, but most frequently occurs after the age of 65. In the early stages, patients and/or their families notice subtle progressive decline in memory, initiative, word-finding, and concentration. As the disease progresses, deficits in memory, language, and reasoning skills become apparent. Some patients also exhibit personality changes and agitation. Depression commonly coexists in patients with dementia. End-stage dementia is characterized by a loss of motor control, incontinence, and mutism. Death typically arises from general lack of mobility, malnutrition, and/or pneumonia.
Promising biomarkers that would allow clinicians to detect evidence of Aβ deposition within the brains of at-risk patients include decreased levels of Aβ42 in the cerebrospinal fluid (CSF) and/or positive amyloid imaging results using positron emission tomography (PET). In 2012 the FDA approved the use of florbetapir for PET amyloid imaging as an adjunct to the evaluation of patients with cognitive impairment. Additionally, biomarkers for neuronal degeneration, including elevated tau in the CSF, PET scans showing decreased glucose metabolism in the temporoparietal cortex, and magnetic resonance imagining (MRI) scans showing advanced cerebral atrophy relative to that of age-matched brains, may be supportive of a probable AD diagnosis.
Screening and Counseling
Less than 5% of AD cases are attributed to autosomal dominant FAD. A handful of rare mutations fully explain the transmission of FAD in select lineages, therefore genetic testing for mutations in APP, PSEN1, and PSEN2 may help to confirm diagnosis. Mutations in the APP and PSEN1 genes explain the majority of familial FAD cases, while mutations in the PSEN2 are significantly rarer with most recorded mutations in this gene arising within a single pedigree known as the Volga Germans.
When carefully monitored, over 50% of aging patients with Down syndrome (DS) show signs of cognitive decline consistent with AD and nearly all brains of DS patients contain substantial numbers of Aβ plaques. The association between AD and DS occurs because the APP gene, which encodes the parent protein of Aβ, is located on chromosome 21. Trisomy 21 therefore causes an overproduction of Aβ presumably leading to AD progression. Family members of AD patients with DS do not have an increased risk for AD.
The APOE genotype is the most robust predictive marker for sporadic AD, however variants of APOE are only associated with risk of developing AD. Those homozygous for the ε4 allele have an 8- to 15-fold increase in risk for AD, while ε4 heterozygotes (individuals with only one copy of ε4 allele) have a 2- to 3-fold increase in risk compared to individuals with the more common ε3/ε3 genotype. Meanwhile, the ε2 allele (the rarest of the APOE risk variants) is thought to be protective for AD. Some studies have also shown that affected ε4 carriers also tend to have an earlier age of onset for AD. While these associations have been most widely studied in Caucasians, the ε4 variant is also associated with AD risk in other ethnicities, but the magnitude of this risk effect varies in different populations. Outside of APOE, discoveries from GWAS have greatly expanded the list of known AD susceptibility genes (Table 124-2). These genes further implicate the compliment system and immune response pathways (perhaps via their involvement in Aβ clearance), mechanisms of endocytosis, and protein trafficking pathways in the pathogenesis of AD. Additionally, a recent sequencing experiment involving over 1700 Icelandic participants has revealed a rare coding mutation in APP that is not only protective for AD, but also appears to be associated with generally reduced symptoms of cognitive decline in aging carriers. While the individual effect size of any one of these non-APOE variants is much smaller than that of APOE, researchers still hope the combined genotypic knowledge of these susceptibility genes may lead to better risk assessments for individuals who seek genetic testing for AD.
| Candidate Gene (Location) | SNP/Variants | ORa [population]a |
|---|



