Type 3 secretion effectors
Abigail Clements, Cedric N. Berger, Mariella Lomma and Gad Frankel, Imperial College London, London, UK
Introduction
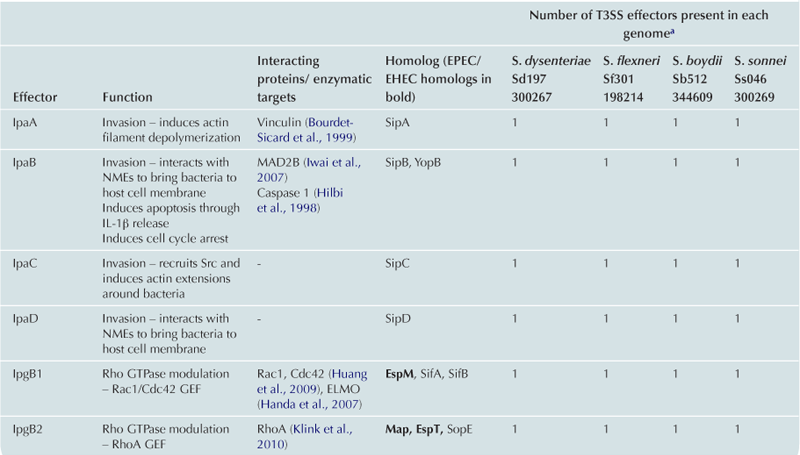
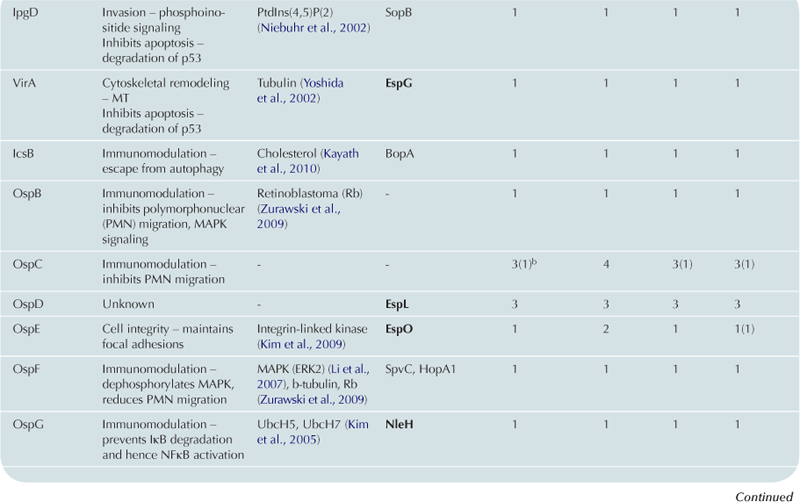
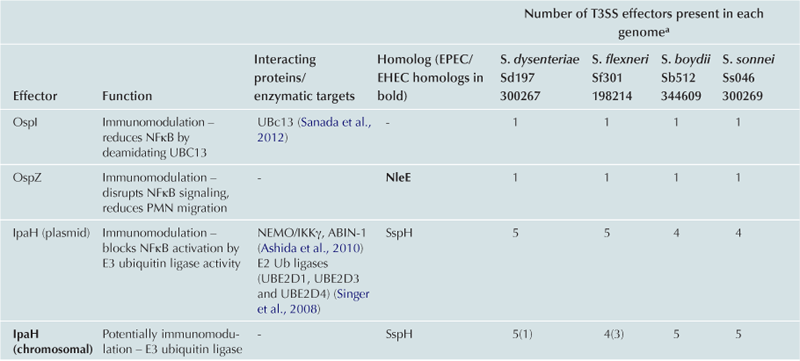
T3SS effector proteins of enterohemorrhagic E. coli (EHEC), enteropathogenic E. coli (EPEC) and Shigella affect diverse signaling pathways and physiological processes when translocated into the host cell. For EPEC and EHEC seven ‘core’ effectors are encoded with the T3S translocation machinery on the locus of enterocyte effacement (LEE) pathogenicity island (PI) (McDaniel et al., 1995), while other effectors are encoded within prophages and other integrative elements (Tobe et al., 2006). Although EPEC and EHEC show high levels of conservation between the LEE encoded effectors, there is significant diversity in their non-LEE effector (NLE) repertoire; EPEC strains E2348/69 and B171 encode at least 23 intact effector genes (Iguchi et al., 2009; Deng et al., 2012), whereas the EHEC O157 strain Sakai is estimated to have closer to 50 T3SS effectors (Table 15.1) (Tobe et al., 2006). The Shigella T3SS is encoded on a large virulence plasmid along with the majority of identified T3SS effectors (Table 15.2). Approximately 30 Shigella T3SS effectors are currently recognized (Parsot, 2009) although as more Shigella isolates of different species are sequenced this number may be revised. This chapter describes how these T3SS effectors modulate the host cytoskeleton, immune response, cell survival, and gut integrity.
TABLE 15.1
T3SS effectors of A/E pathogens
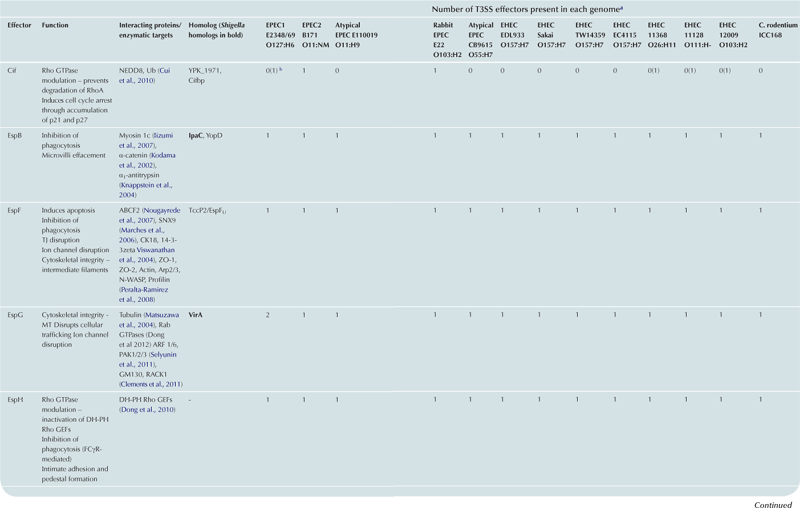
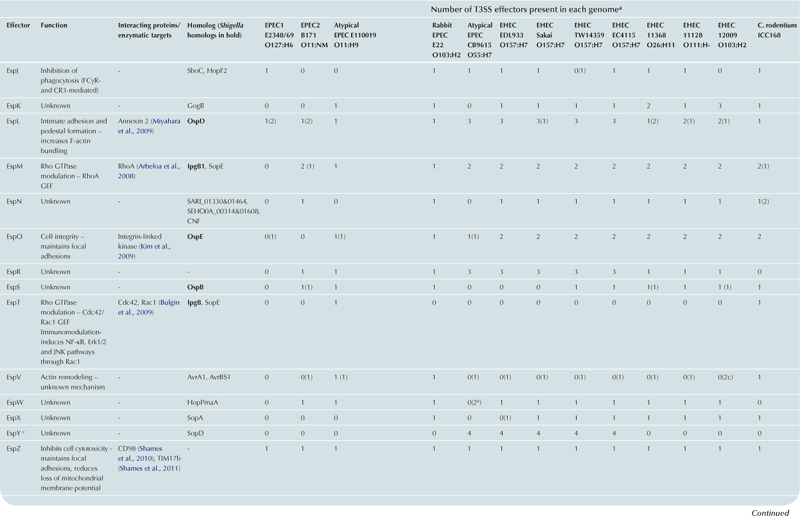
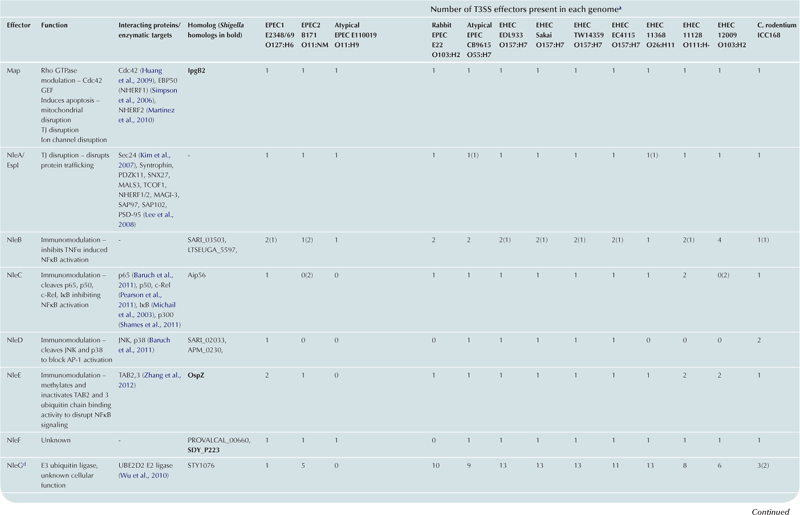
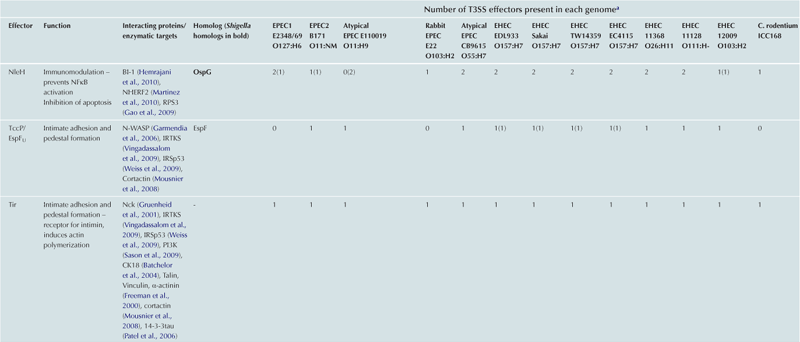
cEspY proteins are grouped by the presence of an N-terminal WEX5F motif. Only homologs of the published EHEC Sakai EspY proteins (Tobe et al., 2006) are indicated and therefore these figures may be an underestimation of the total WEX5F containing effectors.
TABLE 15.2
Cytoskeleton remodeling
EPEC, EHEC and Shigella have acquired a subset of effectors to promote the colonization of the host by modulating the host cytoskeleton. The molecular mechanisms of pedestal formation, cell invasion, modulation of Rho GTPases and reorganization of microtubules and intermediate filaments will be described.
Intimate attachment and pedestal formation
EHEC and EPEC colonization is characterized by the T3SS-dependent formation of attaching and effacing (A/E) lesions on the apical surface of enterocytes. This presents as localized effacement of the microvilli (discussed further below), intimate attachment of the bacteria to the host membrane and actin polymerization underneath the bacterial attachment site. In cultured cells the actin accumulation is visualized as raised pedestal-like structures underneath adherent bacteria. EPEC and EHEC have derived an efficient way to ensure this important step in colonization occurs by expressing an outer-membrane adhesin, intimin, as well as the translocated intimin receptor (Tir), which the bacteria deliver into the host cell via the T3SS. Tir inserts into the host cell membrane in a hairpin loop topology with the extracellular loop interacting with intimin; the bacteria therefore provide both the adhesin and the ‘host cell’ receptor required for attachment (Figure 15.1A).
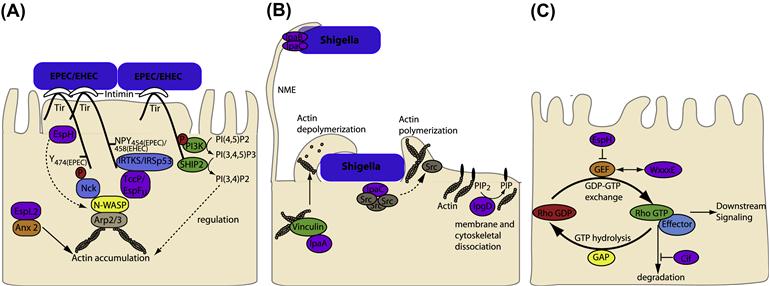
FIGURE 15.1 T3SS effectors from EPEC, EHEC, and Shigella interfere with host cytoskeleton. (A) EPEC and EHEC intimately attach and create pedestals on host cells. The bacterial outer-membrane protein, intimin, interacts with the T3SS effector Tir in the host cell membrane. In some EPEC strains Tir is phosphorylated at Y474 promoting interaction with the host adaptor protein Nck and recruitment of neural Wiskott-Aldrich syndrome protein (N-WASP). EHEC Tir lacks Y474 instead utilizing a NPY458 motif to promote actin polymerization. Tir NPY458 interacts with IRTKS/IRSp53, which binds another T3SS effector TccP/EspFU which in turn recruits N-WASP. In both cases N-WASP recruits the ARP2/3 complex to initiate actin polymerization. N-WASP independent activation of Arp2/3 by TccP/EspFU has also been described and EspH can recruit N-WASP and N-WASP interacting protein (WIP) to the pedestal independent of Tir residues Y474 and NPY458. In addition Tir can interact with phosphoinositide 3-kinase (PI3K) and the inositol-5-phosphatase SHIP2 to regulate actin accumulation in the pedestal through membrane phosphoinositide signaling. EspL2 interacts with Annexin2 (Anx2), increasing aggregation of Tir-induced actin. (B) Nanometer-thin micropodial extensions (NMEs) can interact with the Shigella T3SS effectors IpaB and IpaC and trigger NME retraction bringing the bacteria towards the cell. Invasion occurs through a combination of T3SS effectors; manipulation of Rho GTPases by IpgB1 and 2 (see Figure 15.1C), actin rearrangement through IpaC recruitment of Src, membrane and cytoskeleton dissociation by PIP2 to PIP conversion by IpgD and actin depolymerization by IpaA interaction with vincullin. (C) Rho GTPases cycle between inactive (GDP bound) and active (GTP bound) forms. Rho GEFs activate Rho GTPases by exchanging GDP for GTP and can also participate in binding effector proteins to influence downstream signaling pathways. T3SS effectors from EPEC/EHEC (Map, EspM, and EspT) and Shigella (IpgB1 and IpgB2) mimic Rho GEFs by binding to, and inducing a conformational change in, their respective Rho GTPases to allow GTP binding and activation. The T3SS effector Cif blocks RhoA degradation resulting in stress fiber formation while EspH inactivates one subset of mammalian Rho GEFs.
Intimin is encoded by the eae gene on the LEE PI and is secreted by the general secretory pathway to be inserted into the bacterial outer membrane (McDaniel et al., 1995; Touze et al., 2004). In addition to binding the T3SS effector Tir, intimin can also interact with endogenous host cell proteins, including β1-chain integrins (Isberg and Leong, 1990; Frankel et al., 1996) and nucleolin, which is up-regulated by Shiga toxin production (Robinson et al., 2006).
The T3SS effector Tir not only acts as the host cell receptor for intimin but, upon binding, intimin initiates Tir clustering which mediates protein signaling within epithelial cells. Diverse EPEC and EHEC strains have evolved different mechanisms of Tir signaling, all of which result in actin accumulation underneath the bacterial attachment site (Figure 15.1A). In some EPEC strains, Tir is phosphorylated at tyrosine 474 (Y474p) by host tyrosine kinases, (the specific kinases involved remain unclear) (Phillips et al., 2004; Swimm et al., 2004). Tir phosphorylation promotes its interaction with the SH2 domain of the adaptor protein Nck leading to the recruitment of neural Wiskott-Aldrich syndrome protein (N-WASP) via the Nck SH3 domain (Gruenheid et al., 2001; Phillips et al., 2004). Activation of N-WASP occurs by relieving its autoinhibition fold, allowing interaction with the actin-related protein 2/3 (Arp2/3) complex and initiation of actin polymerization (Gruenheid et al., 2001; Campellone et al., 2002). Proteins normally involved in endocytosis, clathrin (Veiga et al., 2007), CD-2-associated protein (CD2AP) (Guttman et al., 2010), and dynamin-2 (Unsworth et al., 2007), are also involved in pedestal formation via the Nck-dependent pathway, the significance of which requires further investigation. EHEC Tir lacks a tyrosine 474 equivalent and the process of actin polymerization is mediated via the non-LEE encoded T3SS translocated effector protein, TccP (Tir-cytoskeleton coupling protein) (Garmendia et al., 2004) also known as EspFU (E. coli secreted protein F in prophage U) (Campellone et al., 2004). TccP/EspFU interacts with the IRSp53/MIM proteins, IRTKS and IRSp53, which also bind Tir at an Asn-Pro-Tyr (NPY458) tripeptide in the Tir C-terminal domain thereby linking TccP/EspFU indirectly to Tir (Campellone and Leong, 2005; Brady et al., 2007; Vingadassalom et al., 2009). TccP/EspFU also interacts with and activates N-WASP to initiate Arp2/3 recruitment and actin polymerization (Cheng et al., 2008). In mammalian cells TccP/EspFU was also shown to induce Arp2/3 complex-dependent actin polymerization in the absence of N-WASP suggesting that alternative proteins can act in this pathway (Vingadassalom et al., 2010). The NPY motif is conserved in EPEC Tir (as NPY454) but in typical EPEC lineage 1 strains this pathway accounts only for low levels of actin polymerization as these strains do not have a TccP/EspFU homolog (Campellone and Leong, 2005). Both pathways appear to be utilized simultaneously in vitro for most non-O157 EHEC strains, EPEC O119:H6 (Whale et al., 2006) and EPEC lineage 2 strains which carry the homolog TccP2/EspFM (Ogura et al., 2007). The current conundrum is that neither pathway appears to be necessary for A/E lesion formation in vivo in EHEC animal models (infant rabbit and gnotobiotic piglet models (Ritchie et al., 2008) or the C. rodentium murine infection model), or in EPEC and EHEC infection of human intestinal in vitro organ cultures (IVOC) (Schuller et al., 2007; Crepin et al., 2010), indicating that the molecular mechanisms and function of A/E lesion formation during infection are far from understood.
EPEC Tir is also responsible for membrane phosphoinositide signaling as it binds host phosphoinositide 3-kinase (PI3K) at phosphorylated tyrosine 454 (Y454p) (Sason et al., 2009; Selbach et al., 2009) and the inositol-5-phosphatase SHIP2 (Smith et al., 2010). These two enzymes convert PI(4,5)P2 to the predominant membrane phosphoinositide found in wild-type pedestals; PI(3,4)P2. An intermediate, PI(3,4,5)P3, which accumulates when SHIP2 recruitment is prevented, results in multiple elongated pedestals (Smith et al., 2010), suggesting a regulated process of initial PI(3,4,5)P3-enhanced actin polymerization followed by signaling down-regulation by PI(3,4)P2, may occur.
Other T3SS effectors implicated in this initial process of A/E lesions or pedestal formation are EspH, EspB, and EspL. EspH inhibits Rho GTPases activity and as such alters the actin cytoskeleton. In addition to modulating Rho GTPases activity, EspH can also promote N-WASP and WASP interacting protein (WIP) recruitment leading to Arp2/3-mediated actin accumulation, which is independent of Tir residues Y454 and Y474 (i.e. Tir:Nck and Tir:IRTKS/IRSp53 pathways) (Wong et al., 2012) revealing yet another Tir-mediated actin polymerization pathway that may contribute to A/E lesions in vivo.
EspB forms part of the translocation pore at the tip of the T3SS apparatus but in addition is translocated into the host cell where it has an effector function. Ectopic expression of EspB can redistribute host actin in the absence of any other effectors (Taylor et al., 1999) and can bind to host cell proteins including α-catenin (Kodama et al., 2002) and myosins (Iizumi et al., 2007), affecting pedestal formation and initiation of phagocytosis respectively. Both α-catenin and EspB can be seen in the pedestal of EHEC-infected tissue culture cells and are thought to participate in the rearrangement of actin molecules in the pedestal (Hamaguchi et al., 2008). Detailed understanding of EspB’s role in pedestal formation has been hampered by its importance in T3SS pore formation, while the interaction of EspB and myosins has been separated from its pore-forming function, this has not occurred for the interaction of EspB and α-catenin.
EspL2 of EHEC can interact with annexin 2 and increase the ability of annexin 2 to aggregate the Tir-induced actin that accumulates underneath the bacterial attachment site (Miyahara et al., 2009). EspL2 also seems to have a Tir-independent function whereby it induces pseudopod-like protrusions of the host cell membrane to enhance bacterial adherence (Miyahara et al., 2009). EspL proteins are homologs of the Shigella OspD family, however the Shigella OspD proteins do not appear to be involved in actin aggregation (Nataro et al., 1995).
Like its distant homolog TccP/EspFu, EspF possesses N-WASP binding sites, which have been shown in vitro to be functional (Alto et al., 2007). EspF also binds to sorting nexin 9 (SNX9) and induces membrane remodeling and in vitro can nucleate a multiprotein complex of both N-WASP and SNX9 to potentially coordinate membrane remodeling and actin polymerization (Alto et al., 2007).
Invasion and intracellular spread
The uptake of Shigella by the host cell depends on a complex rearrangement of the host cell membrane and cytoskeleton, which is initially triggered by receptor binding but requires the T3SS effectors for complete uptake (Figure 15.1B). Recently it has been shown that bacteria can also be captured by filopodial extensions from the host cell, termed nanometer-thin micropodial extensions (NMEs), and this interaction occurs through the T3SS tip complex proteins IpaB and IpaD (Romero et al., 2011). Connexin-mediated signaling and extracellular ATP stimulates Erk1/2 activation, which controls actin retrograde flow in NMEs resulting in NME retraction, bringing the bacterium into contact with the cell body to allow invasion to occur.
Bacterial invasion begins with the formation of membrane ruffles, which require modulation of Rho GTPase activity by IpgB1 and 2. Rho GTPase modulation is discussed in detail elsewhere in this chapter. In addition, the carboxylterminal domain of IpaC induces actin polymerization responsible for the formation of cell extensions that engulf the bacterium (Tran Van Nhieu et al., 1999; Kueltzo et al., 2003). IpaC appears to do so by recruiting and activating Src tyrosine kinase (Mounier et al., 2009). IpaC also forms part of the T3SS pore and is thus necessary for delivery of T3SS effector proteins into the host cell. A Shigella strain mutated in the IpaC C-terminal domain was proficient in translocating T3SS effector proteins but was unable to recruit Src, uncoupling these two functions of IpaC (Mounier et al., 2009). Src appears to be initially recruited to the intimate contact site of the bacteria but quickly diffuses in an actin-dependent manner into extensions that surround the bacteria (Dumenil et al., 1998), the molecular details governing this process remain to be elucidated.
The T3SS effector IpgD dephosphorylates phosphatidylinositol-4,5-biphosphate [PI(4,5)P(2)] into phosphatidylinositol-5-phosphate [PI(5)P] (Niebuhr et al., 2002). This dephosphorylation event leads to a decrease in membrane tether force, which can be seen by increased membrane blebbing and actin filament remodeling upon ectopic expression of the protein (Niebuhr et al., 2002). During infection this may permit localized dissociation between the membrane and cytoskeleton to allow membrane ruffling and filopodia extension through the manipulation of Rho GTPases by IpgB1 and IpgB2. The IpaA protein binds to the focal adhesion protein vinculin (Tran Van Nhieu et al., 1997), increasing the association of vinculin with F-actin and inducing actin filament depolymerization (Bourdet-Sicard et al., 1999), which may be required for completion of the entry process or for reappropriating actin.
In Shigella, actin-based motility occurs through the action of IcsA, an autotransporter rather than a T3SS effector, which recruits and activates N-WASP, Arp2/3, polymerizing actin.
Rho GTPase modulation
Many T3SS effectors subvert host cell actin dynamics by disrupting Rho GTPase signaling (Bulgin et al., 2010). The Rho family GTPases are crucial in the regulation of key cellular functions and the best characterized members are Cdc42, Rac1, and RhoA, which trigger filopodia, lamelipodia/ruffles, and stress fibers respectively (Hall, 1998). Rho GTPases cycle between an active, GTP-bound state (predominantly membrane associated) and an inactive GDP-bound form (predominantly cytoplasmic). As shown in Figure 15.1C, the exchange of GDP for GTP is stimulated by guanine exchange factors (GEFs) to activate Rho GTPases. Some RhoGEFs can also mediate interaction with effector molecules to determine downstream signaling pathways. GTP hydrolysis is enhanced by binding of GTPase-activating proteins (GAPs) resulting in inactive GDP-bound Rho GTPase, again some GAPs can scaffold protein complexes. In addition, GDP release is blocked by guanine nucleotide-dissociation inhibitors (GDIs) maintaining the inactive state of Rho GTPases in the cytosol. Therefore these regulators of Rho GTPases not only define the Rho GTPase activity of a molecule but also influence the formation of multiprotein complexes and hence downstream signaling events.
A number of bacterial effector proteins have been described which modulate Rho GTPases and many were grouped on the basis of a WxxxE motif and suggested to act as Rho GTPase mimics (Alto et al., 2006). Structural information helped establish that representatives of this family of effectors actually act as RhoGEFs by binding to their respective Rho GTPases and inducing a conformational change to allow GTP binding and hence activation (Buchwald et al., 2002; Ohlson et al., 2008). These include the EPEC/EHEC effectors Map (Cdc42 GEF[53]), EspM (RhoAGEF[54]), and EspT[55] (likely Rac1 and Cdc42 GEF, although direct binding has not been demonstrated) (Bulgin et al., 2009) and the Shigella effectors IpgB1 (a Rac1 and Cdc42 GEF) and IpgB2 (a RhoA GEF) (Huang et al., 2009; Klink et al., 2010).
Activation of Cdc42 by Map results in localized transient filopodia formation (Kenny et al., 2002; Berger et al., 2009). Map can polarize Cdc42 at the cell membrane to form actin-rich protrusions in the absence of external stimuli. This reaction is reliant on the interaction of Map with EBP50 (NHERF1), which interacts with Ezrin to link Map to the actin cytoskeleton (Orchard et al., 2012). During infection it is hypothesized that localized actin rearrangement at the bacterial attachment site is recognized by the Map-EBP50-ezrin complex, which recruits Cdc42 to the attachment site leading to further actin polymerization. EspM activation of RhoA results in stress fiber formation in a ROCK-dependent manner (Arbeloa et al., 2008), while EspT induces formation of membrane ruffles (through the Rac1 effector Wave2), lamellipodia, and bacterial internalization (Bulgin et al., 2009).
The Shigella Rho GEF IpgB1 associates with the plasma membrane and was previously shown to recruit the ELMO-Dock180 complex to the membrane to activate Rac1 (Handa et al., 2007), whether this occurs in addition to its RhoGEF activity remains to be confirmed. IpgB1, but not IpgB2, is necessary for efficient invasion of HeLa cells (Ohya et al., 2005; Hachani et al., 2008). However in polarized cells only a double IpgB1/B2 mutant showed significantly reduced invasion (Hachani et al., 2008) and therefore the interplay between these effectors in triggering host cell invasion requires further investigation.
No T3SS GAPs or GDIs have been identified to date in EPEC/EHEC or Shigella, however other T3SS effectors can modulate Rho GTPases in different ways. The EPEC effector Cif stabilizes RhoA (Cui et al., 2010) resulting in stress fiber formation (Oswald et al., 1994). Cif deamidates the ubiquitin-like protein NEDD8, which in turn deactivates neddylated Cullin-RING ubiquitin ligases, a substrate of which is RhoA. RhoA is therefore not ubiquitinated and degraded when Cif is present (Cui et al., 2010). The EPEC/EHEC effector EspH subverts actin dynamics affecting filopodia and pedestal formation (Tu et al., 2003). EspH inactivates mammalian RhoGEFs which contain the Dbl-homology and pleckstrin-homology (DH-PH) domains, but does not inactivate the bacterial RhoGEFs (Wong et al., 2012). This poses an interesting scenario where EspH may potentially reduce a subset of the endogenous mammalian RhoGEFs in order for the bacterial RhoGEFs to take over cell signaling for the benefit of the bacteria.
T3SS effectors can also modulate Rho GTPase signaling pathways by binding directly to the Rho GTPase effector, bypassing the need for Rho GTPase activation. EspG and TccP/EspFU are examples of such effectors (Selyunin and Alto, 2011). Rho GTPase effectors such as N-WASP and p21 activated kinases (PAKs) have autoinhibitory Rho GTPase binding domains (GBD) which normally inhibit the activity-bearing domain (AD). For WASP the AD is a VCA domain (verprolin homology, central hydrophobic and acidic regions) which recruits the Arp2/3 complex while the PAK AD is a kinase domain. TccP/EspFU binds to the N-WASP GBD releasing the autoinhibition of N-WASP allowing it to recruit Arp2/3 and nucleate actin (Cheng et al., 2008), as described above. EspG binds to PAK releasing the kinase domain from autoinhibition, although to date this has only been demonstrated in vitro (Selyunin et al., 2011). The method of release and activation of the autoinhibited AD by these bacterial effectors is different to the mechanism by which endogenous Rho GTPases activate their effectors, indicating the bacterial effectors have developed a novel way to manipulate these host proteins.
Other cytoskeletal components (MT and IF)
The T3SS effector VirA was initially identified as being required for the efficient entry of bacteria into epithelial cells (Uchiya et al., 1995) and was subsequently demonstrated to be able to destabilize microtubules producing membrane ruffles and Rac1 activation (Yoshida et al., 2002). Further investigation suggested VirA had protease activity and could selectively degrade α-tubulin allowing intracellular and intercellular spreading (Yoshida et al., 2006). Recently VirA was shown not to directly degrade tubulin or microtubules (Davis et al., 2008; Germane et al., 2008) and the mechanism of microtubule disruption remains unclear. The VirA homolog EspG from EPEC and EHEC was also shown to induce microtubule disruption, releasing GEF-H1 from the cytoskeleton and activating RhoA resulting in actin stress fiber formation (Matsuzawa et al., 2004). New evidence suggests EspG acts as a Rab GTPase activating protein (RabGAP) trapping Rab GTPases in their inactive GDP bound form (Dong et al. 2012), in addition to binding PAKs and ARF GTPases (Selyunin et al., 2011), all of which may have downstream effects that result in cytoskeletal rearrangements.
EspF interacts with cytokeratin 18 (CK18), a protein that forms part of the intermediate filament network in epithelial cells and can be seen recruited to the site of bacterial attachment (Batchelor et al., 2004). CK18 can also bind Tir (Batchelor et al., 2004) and the adapter protein 14-3-3 (Patel et al., 2006), which EspF and Tir can also bind (Viswanathan et al., 2004). These interactions potentially allow Tir and EspF to coordinate collapse of the intermediate filament network with actin redistribution and polymerization.
Manipulation of host immune responses
Manipulation of the host immune system is a common theme in bacterial infections and a requirement for successful colonization and dissemination. Bacteria must evade phagocytosis, modulate cell-intrinsic innate immunity and avoid autophagy in order to survive and proliferate.
Inhibition of phagocytosis
The first line of host immune defenses involves professional cells such as macrophages, neutrophils, and dendritic cells, which internalize and destroy bacteria and other invaders. Phagocytosis is a process by which phagocytic cells internalize particulate material and is therefore distinct from other forms of endocytosis such as the vesicular uptake of fluids. Phagocytosis is a multistep process triggered by the recognition of specific ligands by surface receptors and local remodeling of the actin cytoskeleton. Well-characterized phagocytic pathways involve Fc gamma receptor (FcγR) and complement receptor 3 (CR3) that bind IgG and C3bi respectively (Caron and Hall, 1998). Importantly EPEC and EHEC colonize gut epithelium but remain predominantly extracellular. Indeed EPEC is able not only to block its own uptake by professional phagocytes (cis-phagocytosis), but is able also to inhibit the phagocytosis of IgG-opsonized particles (trans-phagocytosis) (Goosney et al., 1999; Celli et al., 2001; Quitard et al., 2006). Inhibition of both cis- and trans-phagocytosis is T3SS dependent.
The first T3SS effector to be identified as responsible for inhibition of phagocytosis was EspF (Quitard et al., 2006; Marches et al., 2008). EPEC espF mutants are phagocytosed by mouse-derived macrophages to the same extent as EPEC lacking a functional T3SS (Quitard et al., 2006; Marches et al., 2008). The role of EspF in inhibition of phagocytosis seems to be correlated with its ability to inhibit PI3K (Quitard et al., 2006). The antiphagocytic activity of EspF is due to the N-terminal region that also contains binding sites for actin, profilin, and SNX9 (Quitard et al., 2006; Alto et al., 2007; Peralta-Ramirez et al., 2008). These partner proteins have not been shown to be involved in antiphagocytosis, but as a specific mechanism for EspF activity still remains to be elucidated, their involvement cannot be discounted. EspF was shown to block only cis- and FcγR-dependent phagocytosis (Marches et al., 2008).
EspB binds the actin-binding domain of multiple myosin family members (Iizumi et al., 2007). Myosins (myosin-1c, -2, -5, -6, and -10) are involved in phagocytosis as they localize at the phagocytic cup and their inhibition suppresses phagocytocis (Swanson et al., 1999). Furthermore, early reports have shown that tropomysin is recruited at the site of EPEC adherence (Goosney et al., 2001). Through its interaction with myosins EspB can compete with actin to bind myosins at the actin-binding domain and may then interfere with myosin-induced phagosome constriction. An internal region of EspB (amino acids 159–218) was identified as responsible for this interaction (Iizumi et al., 2007) and deletion of this region reduced the ability of EPEC to inhibit phagocytosis but was not required for bacterial adherence or delivery of T3SS effectors pointing to a specific role in antiphagocytosis (Iizumi et al., 2007).
A recently identified effector involved in inhibition of phagocytosis is EspH, which blocks the activation of Rho GTPases by binding DH-PH Rho GEFs (Dong et al., 2010). The impact of EspH on actin cytoskeleton dynamics is reflected at the level of phagocytosis as Rho GTPases control cytoskeleton remodeling during FcγR-dependent phagocytosis (Caron and Hall, 1998) and EPEC espH mutants have a reduced ability to inhibit FcγR-dependent phagocytosis (Dong et al., 2010).
Uniquely, EspJ alone can inhibit both FcγR- and CR3-dependent phagocytosis (Marches et al., 2008). EspJ expression appears to only block phagocytosis of opsonized particles (trans-phagocytosis) but not self-uptake (cis-phagocytosis). Similarly to EspF, translocated EspJ localizes to mitochondria (Kurushima et al., 2010) but this does not preclude a role for EspJ at the phagocytic cup. The mechanism by which EspJ blocks phagocytosis is currently unknown.
Subversion of innate immunity
In mammals a complex set of signaling networks initiate both innate and adaptive immune responses against bacterial pathogens. Pathogens have therefore evolved sophisticated mechanisms to subvert these signaling networks in order to circumvent host innate immune responses.
EPEC infection in vivo results in intestinal tissue damage, neutrophil infiltration of the infected mucosa and damage of the gut epithelium (Savkovic et al., 1996; Chakravortty and Kumar, 1999; Michail et al., 2003). This pathology has been linked to the inflammatory responses mounted by the cells of the infected tissues. EPEC and EHEC interaction with the gut mucosa results in the stimulation of pattern recognition receptors (PRRs), such as toll-like receptors (TLRs), that recognize specific pathogen-associated molecular patterns (PAMPs), including flagellin and lipopolysaccharide (LPS) (Khan et al., 2006; Schuller et al., 2009). TLRs initiate signaling pathways downstream of these receptors ultimately converging on a set of transcriptional activators including interferon-regulatory factors (IRFs), nuclear factor-κB (NF-κB), and mitogen-activated protein kinases (MAPKs) resulting in the expression and secretion of pro-inflammatory chemokines and cytokines such as interleukin 8 (IL-8), IL-6, IL-12, and tumor necrosis factor α (TNFα).
The pathway best described in the case of EPEC infection is the NF-κB pathway. The NF-κB family comprises five proteins that can form homo- and/or heterodimeric complexes: RelA (p65), RelB, c-Rel, p50 (NF-κB1), and p52 (NF-κB2), but the most abundant form in mammalian tissues is the dimer p50/p65 (Li and Verma, 2002). Under non-stimulating conditions, NF-κB is kept inactive in the cytoplasm through its association with inhibitory proteins (IκBs), this interaction masks the nuclear localization signal (NLS) of p65 thus preventing nuclear translocation. Molecular mechanisms that lead to NF-κB activation have been studied intensively. Briefly, ligand-bound TLRs recruit the adaptor protein MyD88, which in turn recruits the IL-1R-associated kinases 1 (IRAK1) and 4 (IRAK4), leading to their sequential autophosphorylation and activation (Silverman and Maniatis, 2001). Phosphorylated IRAK1 then associates with TNF receptor-associated factor 6 (TRAF6). This association activates the E3 ubiquitin ligase function of TRAF6, which, with the UBC13/UEV1 E2 ubiquitin-conjugating complex, catalyzes the synthesis of Lys-63-linked polyubiquitin chains (Deng et al., 2000). These ubiquitin chains serve as a scaffold to recruit both the transforming-growth-factor-β-activated kinase 1 (TAK1) and IκB kinase complexes (IKK) through their respective ubiquitin-binding subunits, TAK-binding protein 2 and 3 (TAB2/3) and NF-κB essential modulator (NEMO) (Kanayama et al., 2004; Ea et al., 2006; Wu et al., 2006). As a result of their proximity, TAK1 can phosphorylate the IKK-β subunit of IKK, which then phosphorylates IκBα. Release of NF-κB dimers occurs after phosphorylation of IκB which allows the ubiquitination by SCFβTrCP complex and subsequent proteasomal degradation of IκBα, allowing NF-κB translocation into the nucleus. Here, NF-κB binds specific DNA consensus sequences and activates expression of pro-inflammatory cytokines.
EPEC initially activates the NF-κB signaling pathway through a T3SS-independent mechanism, and subsequently utilizes a T3SS-dependent mechanism to inhibit NF-κB activation and production of pro-inflammatory cytokines (Hauf and Chakraborty, 2003; Maresca et al., 2005) (Figure 15.2A). In recent years an increasing number of studies have demonstrated that EPEC and EHEC encode and translocate into cells a set of protein effectors that target specific regulators of the NF-κB pathway to disarm the host inflammatory response (Nadler et al., 2010; Newton et al., 2010; Vossenkamper et al., 2010; Yen et al., 2010; Baruch et al., 2011; Muhlen et al., 2011; Pearson et al., 2011; Zhang et al., 2012).
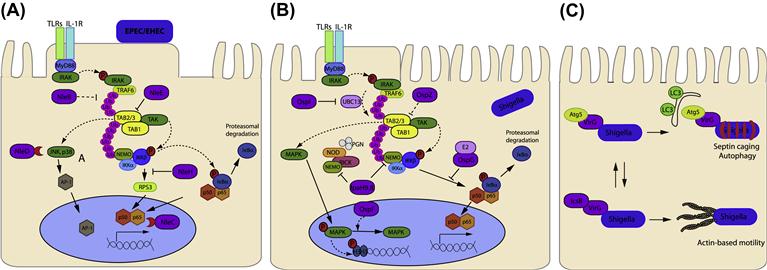
NleE from EPEC/EHEC and OspZ from Shigella (Zurawski et al., 2008) belong to the same family and play a key role in modulating the innate immune response during infection by blocking the translocation of activated NF-κB (p65 and c-Rel but not p50) to the nucleus, thereby decreasing the expression and production of IL-8 (Nadler et al., 2010; Newton et al., 2010; Zhang et al., 2012). This effect is seen in both epithelial cells and dendritic cells (Vossenkamper et al., 2010). NleE prevents IκB degradation in response to TNFα or IL-1β suggesting that the effector targets components shared by these inflammatory signaling pathways.
NleE has a unique S-adenosyl-L-methionine-dependent methyltransferase (SAM) activity that specifically modifies a zinc-coordinating cysteine in the Npl4 zinc finger (NZF) domains in TAB2 and TAB3. This cysteine methylation disrupts ubiquitin chain binding of TAB2/3 and blocks TRAF6-induced activation of TAK1 (Zhang et al., 2012). A 6-aa motif IDSYMK209–214 has been shown to be critical for the immunosuppressive function of NleE (Newton et al., 2010). Indeed, the crystal structure of NleE revealed a SAM-binding pocket, which involves Y212 and R107. Mutation of either residue abolished NleE methylation of TAB3-NZF (Zhang et al., 2012). Given the abundance of biological processes relying on the activation of zinc-finger motifs by cysteine-zinc binding, specific methylation of zinc-coordinating cysteine residues might regulate other eukaryotic pathways in addition to NF-κB signaling.
Another effector involved in the stabilization of IκB is the highly conserved effector NleB. NleB1 from EPEC inhibits NF-κB activation as effectively as NleE in response to TNFα but not IL-1β suggesting that NleB1 may act at a different point of the NF-κB signaling pathway and independently from MyD88/IRAK activation of TRAF6 (Kelly et al., 2006; Petty et al., 2010). The mechanism, however, is still unknown.
The NF-κB pathway is also targeted by the effector NleC. This effector contains a consensus zinc metalloprotease motif 183HEIIH187 that is responsible for its proteolytic activity. Indeed NleC was recently described as a potent protease of EPEC that cleaves and inactivates the NF-κB p65 subunit (Petty et al., 2010; Yen et al., 2010; Baruch et al., 2011; Muhlen et al., 2011; Pearson et al., 2011). Site-directed mutagenesis of the histidines within the consensus sequence is sufficient to abrogate NleC proteolytic activity (Petty et al., 2010; Yen et al., 2010; Baruch et al., 2011; Muhlen et al., 2011; Pearson et al., 2011). Furthermore, NleC cleaves p65 within its conserved DNA-binding domain, suggesting that other NF-κB family members containing the same sequence could be substrates of NleC (Baruch et al., 2011). The acetyltransferase p300 was recently identified as an additional target of NleC (Shames et al., 2011). p300 functions as a co-activator in the transcription of many genes by acetylating p65 (among other transcription factors), thereby enhancing expression of genes downstream of κB-containing enhancers (Chen et al., 2001) such as IL-8. Overexpression of p300 can indeed antagonize repression of IL-8 secretion induced by EPEC infection (Shames et al., 2011). While infection with EPEC nleC mutant does not result in increased IL-8 production, presumably due to functional redundancy with NleE and NleB, infection with an EPEC nleEC double mutant or an EPEC nleBEC triple mutant results in significantly higher IL-8 production by host cells compared with infection with EPEC nleE or EPEC nleBE alone (Baruch et al., 2011; Pearson et al., 2011). Thus, NleC appears to complement the activities of NleE and NleB by eliminating activated p65/p50 and/or its acetyltransferase p300 that would positively influence transcription and expression of pro-inflammatory cytokines.
Together with NleC, the effector NleD was identified as zinc-dependent metalloprotease as it also harbors the consensus sequence (142HELLH146) (Marches et al., 2005). NleD specifically targets MAP kinases JNK and p38 but not ERK, thereby blocking nuclear translocation of the AP-1 transcription factor, which regulates multiple cell processes including inflammation, differentiation, proliferation, and apoptosis (Baruch et al., 2011). Infection with an EPEC nleBECD mutant results in an increase of expression and secretion of IL-8 compared to the nleBEC mutant, indicating an anti-inflammatory role for NleD (Baruch et al., 2011).
The NleH family of proteins is also conserved amongst EPEC and EHEC strains and the C-termini of NleH1 and NleH2 proteins display significant amino acid sequence similarity with the S. flexneri T3SS anti-inflammatory effector OspG (Kim et al., 2005). NleH1 and NleH2 have critical roles in inhibiting host cell apoptosis; however, these effectors have also been implicated in the suppression of NF-κB activation. NleH1 and NleH2 decrease IκKβ-induced NF-κB activity by decreasing TNFα-induced degradation by preventing ubiquitination of phospho-IκBα. Although the mechanism is not clear yet, stabilization of IκBα is dependent on conserved lysine residues K159 in NleH1 and K169 in NleH2 from EPEC E2348/69 respectively (Royan et al., 2010). It was recently shown that the N-terminus of both NleH1 and NleH2 could bind the ribosomal protein S3 (RPS3) (Gao et al., 2009). RPS3 is a non-Rel subunit of NF-κB complexes (Wan et al., 2007). RPS3 is considered a ‘specifier’ subunit of NF-κB, because it facilitates high-affinity binding of DNA and determines the specificity of NF-κB for selected target genes (Wan et al., 2007). NleH1 of EHEC selectively blocks the transcription of NF-κB target genes by attenuating nuclear translocation of RPS3 without affecting p65 localization (Gao et al., 2009). Nuclear translocation of RPS3 is triggered by IκKβ kinase-dependent phosphorylation at S209, which enhances RPS3 association with nuclear importin-α. NleH1 specifically inhibits IκKβ-dependent phosphorylation of S209 thus blocking RPS3 function (Wan et al., 2011). Interestingly, another crucial target of IκKβ phosphorylation in the NF-κB pathway is IκBα but whether NleH can block IκKβ-dependent phosphorylation of IκBα remains to be elucidated as well as the mechanism employed by NleH to block such post-translational modification.
By translocating into the host cell cytoplasm effectors such as NleE, NleB, NleC, NleD, and NleH, EPEC and EHEC employ a highly coordinated strategy to subvert inflammatory signaling pathways. This coordinated attack allows the bacteria to establish infection and avoid rapid clearance by the host innate immune response.
Similar to EPEC and EHEC Shigella delivers a subset of effectors via its T3SS to subvert cellular and immune functions to promote infection (Figure 15.2B). During invasion and multiplication within the cells of the intestinal epithelium Shigella peptidoglycan stimulates the intracellular PRR receptor Nod-like receptor 1 (NOD1), which interacts with the serine-threonine kinase RICK (also called Ripk2 or RIP2) to induce NF-κB and MAPK p38, ERK, and JNK signaling (Kanneganti et al., 2007). Both MAPK and NF-κB pathways components are targeted by Shigella T3SS–delivered effectors (Ashida et al., 2011).
As mentioned above, among its repertoire of T3SS effectors Shigella translocates OspZ and OspG, homologs of NleE and NleH respectively. Similar to NleE, OspZ blocks the translocation of NF-κB to the nucleus but whether OspZ from Shigella shares the same cysteine methylase activity of EPEC NleE is not currently known (Zurawski et al., 2008; Newton et al., 2010).
A yeast two-hybrid screen showed that OspG is able to bind several ubiquitinated E2 Ub-conjugating enzymes, including UbcH5 (Kim et al., 2005). The interaction with UbcH5 seems to inhibit E2 activity, which is indispensable for the proper activation of the E3 Ub ligase SCFβTrCP, the specific SCF complex that controls IκBα degradation. Therefore, OspG negatively regulates the NF-κB inflammatory response by interfering with the proteasome-dependent degradation of IκBα. OspG also displays kinase activity and induces its autophosphorylation. This activity appears to be necessary for the function of OspG, but does not seem to be involved in the phosphorylation of IκBα before ubiquitination and proteasome-mediated degradation. Consistent with the OspG inhibiting SCFβTrCPactivity, epithelial cells infected with S. flexneri wild-type strain, but not an ospG mutant, accumulate phosphorylated IκBα and the ospG mutant induced a much stronger inflammatory response during infection in vivo than the wild-type strain (Kim et al., 2005).
IpaH9.8 is another effector delivered by Shigella into host cells that interferes with the innate immune response. Once injected into the host cell IpaH9.8 translocates to the nucleus (Toyotome et al., 2001) and has been shown to be an E3 Ub ligase (Rohde et al., 2007). In vitro, IpaH9.8 displayed ubiquitin ligase activity specific for the yeast MAPK Ste7. Indeed, in yeast cells IpaH9.8 interrupts pheromone response signaling by promoting the proteasome-dependent destruction of Ste7 (Rohde et al., 2007) and replacement of an invariant Cys residue abolished the ubiquitin ligase activity of IpaH9.8 (Rohde et al., 2007). More recently IpaH9.8 has been shown to dampen the host NF-κB-mediated inflammatory response in mammalian cells (Ashida et al., 2010). IpaH9.8 interacts directly with NEMO/IKKγ and ABIN-1, an ubiquitin adaptor protein, promoting ABIN-1-dependent polyubiquitination of NEMO on two specific Lys residues (K309 and K321). Consequently, polyubiquitinated NEMO undergoes proteasome-dependent degradation and blocks NF-κB activation (Ashida et al., 2010).
OspF, another T3SS effector of Shigella, is translocated into the host cell nucleus, where it targets components of the MAPK pathway (Arbibe et al., 2007). OspF has a phosphothreonine lyase activity, which mediates irreversible dephosphorylation of MAPKs thereby inactivating them. This irreversible modification inhibits the downstream phosphorylation of histone H3 at Ser10 and ultimately results in the inhibition of transcriptional activation of NF-κB-regulated genes, including IL-8 (Arbibe et al., 2007; Kramer et al., 2007; Li et al., 2007; Zurawski et al., 2009).
OspI was recently identified as a new T3SS effector of Shigella that selectively deamidates a glutamine residue to glutamic acid in the E2 enzyme UBC13. This modification abolishes UBC13 E2 ubiquitin-conjugating activity required for the activation and polyubiquitylation of TRAF6. The authors also demonstrated that through OspI activity, Shigella can block NF-κB-mediated inflammatory response at an early stage of epithelial invasion (Sanada et al., 2012). Indeed, OspI encoding gene (orf169b) was initially identified because its mutation led to a dramatic increase in the expression of pro-inflammatory chemokines and cytokines in HeLa-infected cells at early stages of infection. Although the impact of OspI activity on NF-κB pathway was clearly demonstrated, it still remains to be elucidated whether by abolishing TRAF6 activation OspI can also influence MAPKs mediated signaling pathways.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


