Urinary Tract Infection
There are two main clinical syndromes:
Bacteriuria is confirmed by finding a urinary excretion of more than 100,000 organisms/ml urine (counts of <10,000/ml are usually caused by contamination). Infection may be symptom-free. Escherichia coli is the most frequent organism (70–80% of cases). Other organisms (Proteus, Staphylococcus, Streptococcus, Klebsiella and Pseudomonas) are usually associated with structural abnormality or catheterisation, and reinfection. Tuberculosis classically causes a sterile pyuria. Pyuria can almost always be detected by careful microscopic examination of fresh unspun urine. Microscopic haematuria is common.
Management
Uncomplicated cases are treated with oral antibiotics such as trimethoprim and ampicillin (3-day course for cystitis, at least 7 days for pyelonephritis) after obtaining urine for culture and antibiotic sensitivity. Resistant organisms may be sensitive to co-amoxiclav (Augmentin) or ciprofloxacin. Patients with acute pyelonephritis who are vomiting or have evidence of septicaemia (blood cultures are positive in 20% of cases) require intravenous antibiotics.
There may be an obvious predisposing cause, e.g. pregnancy, urinary obstruction or catheterisation. Diabetes mellitus must be excluded. Acute pyelonephritis or more than two episodes of cystitis in a woman, or any infection in a man, suggest a structural abnormality. Ultrasound of the renal tract is performed to look for perinephric abscess, renal scarring, stone, tumours or obstruction. CT scanning or intravenous urography (IVU) and possibly cystoscopy may be necessary (frequent infections, persistent haematuria, dysuria or loin pain) to exclude small stones/tumours or bladder diverticula.
Women prone to recurrent infections should be given advice about complete emptying of the bladder (double micturition) and voiding soon after intercourse. Low-dose antibiotic prophylaxis (e.g. trimethoprim or cephalexin) reduces the incidence of infection and can be used safely for long periods.
Children require investigation as infection in the presence of vesicoureteric reflux can lead to permanent kidney damage.
Imaging the Kidneys
A plain abdominal film usually shows the renal outlines and identifies any calcification in the renal tract.
Renal ultrasound is useful in determining renal size and contour, and defining the size, location and consistency (solid or cystic) of any renal mass, and looking for pelvicalyceal dilatation of obstruction.
IVU has the advantage of demonstrating the whole urinary tract, but its usefulness has been replaced in many cases by ultrasound, computed tomographic (CT) scanning and radionuclide scanning. Ultrasound and CT are particularly useful for anatomical studies, and radionuclide scanning for providing functional information. IVU should not be performed if there is a history of sensitivity to contrast media. Dehydration prior to the examination should be avoided in renal failure, diabetes or myeloma.
Isotope scanning (most commonly 99mTc-diethylenetriaminepentacetic acid (DTPA) or 99mTc-dimercaptosuccinate (DMSA)) can be used to assess renal blood flow, renal function and transit time of filtrate across the parenchyma into the collecting system. It can be useful in the diagnosis of renal artery stenosis and obstruction. In addition, the renal parenchyma can be visualised for evidence of scarring.
Stones
Eighty percent of urinary tract stones contain calcium, usually as calcium oxalate. Less common constituents are uric acid (10%) or cystine. Staghorn calculi contain struvite, made up of calcium, ammonium and phosphate. Classical features are severe loin pain, with microscopic or macroscopic haematuria.
Clinical Features
The most common presentation is with severe loin pain radiating to the groin (renal colic), with microscopic or macroscopic haematuria. About 1 in 1000 men and 1 in 3000 women present with their first kidney stone in a single year. Fifteen percent of patients develop recurrent stones within a year of first presentation, 30% by 5 years.
Recurrent stones should be investigated for a metabolic cause:
- hypercalciuria – 50% of stone-formers have increased urinary calcium excretion
- elevated serum calcium – usually caused by hyperparathyroidism in stone-formers
- hyperuricaemia
- cystinuria.
Management
The diagnosis is confirmed by imaging. Abdominal X-ray may detect calcium-containing stones. Ultrasound usually identifies stones and will detect dilatation of the renal pelvis or ureter, indicating obstruction. IVU or CT scanning provide the most sensitive methods of detecting stones. Most small stones (< 4 mm) will pass spontaneously, but those > 6 mm are rarely passed. In such cases stones are cleared by extracorporeal shock wave lithotripsy, endoscopic removal, either percutaneously or through cystoscopy with retrograde urethroscopy, or open surgical procedure.
Measures to Prevent Stone Formation
- Increased fluid intake – at least 2 l/day.
- Diet – increased risk of stone formation is associated with low rather than high calcium diet, and with diets high in sodium and protein.
- Thiazide diuretics reduce urinary calcium in hypercalciuria.
- Allopurinol reduces urinary uric acid excretion.
- Penicillamine and captopril form a complex with cystine, which renders it more soluble, and can be used to prevent or dissolve stones.
- Alkalinisation of urine increases solubility of uric acid and cystine and may be of value in preventing uric acid or cystine stone formation by increasing solubility of these compounds.
Chronic Interstitial Nephritis
The term chronic pyelonephritis, which implies infection, has been replaced by chronic interstitial nephritis, which is characterised by a chronic tubulointerstitial inflammatory infiltrate. Interstitial involvement is usually secondary to papillary or tubular damage by infection, ischaemia, radiation, toxins or metabolic disease. The most common cause is reflux nephropathy (see below). Other causes include obstructive uropathy, drugs (cyclosporin, lithium, chronic analgesic ingestion), renovascular disease, sickle-cell disease, long-standing hypokalaemia, hypercalcaemia or hyperuricaema, tuberculosis, sarcoid, heavy metal poisoning (lead, cadmium), radiation nephritis, Sjögren syndrome and hereditary nephritides (e.g. Alport syndrome).
Clinical Features
There is usually altered tubular function (glycosuria, aminoaciduria, renal tubular acidosis and tubular proteinuria) with a variable degree of renal failure. Ultrasound and radionuclide scans may show obstruction, and the kidneys are often small and scarred.
Management
Treat any underlying cause. Give antibiotics (prophylactic if necessary) for infection. Patients are commonly unable to concentrate their urine, and need a high fluid intake.
Reflux Nephropathy
Reflux of urine through a congenitally abnormal vesicoureteric junction occurs in about 1% of infants. Reflux of sterile urine into the kidney may cause renal damage through hydrostatic injury, but there is clear evidence that reflux of infected urine leads to renal scarring. Reflux is present in 50% of infants who develop urinary infection during their first year, and one-third of children who have infection before the age of 12 years. Reflux can also present with enuresis, hypertension and proteinuria. There is a familial incidence.
Management
Children with urinary infections (and possibly those with affected siblings or parents) should be screened with an ultrasound of the renal tract followed if necessary by a direct or indirect radionuclide micturating cystogram. Ureteric reimplantation and conservative treatment with antibiotics to prevent infection are equally effective in preventing scarring. Without surgery reflux generally resolves as the child grows older.
Proteinuria
Small amounts of low-molecular-weight proteins are normally filtered by the glomerulus, and reabsorbed or catabolised by proximal tubular cells. The kidneys normally excrete 50–80 mg protein daily, of which 30–50 mg is Tamm–Horsfall protein, a mucoprotein secreted by tubular cells. Proteinuria > 150 mg/day is abnormal, but proteinuria is more commonly quantified as urinary albumin creatinine ratio (ACR) or protein creatinine ratio (PCR), which are more easily obtained on a spot urine sample and tend to be more reproducible than 24 h collections. ACR > 2.5 mg/mmol in men and 3.5 mg/mmol in women or PCR > 15 mg/mmol are abnormal. A PCR of 100 mg/mmol or ACR of 70 mmol/l is approximately equal to 1 g of protein per 24 h. The conversion is non-linear for levels below this. Dipsticks primarily detect albumin and are relatively insensitive at detecting immunoglobulins or Bence Jones protein (immunoglobulin light chains). Microalbuminuria (urinary albumin excretion of 30–300 mg/day) is an early sign of diabetic nephropathy (p. 242).
Causes
- glomerular disease: glomerulonephritis, glomerulosclerosis (diabetic and hypertensive), glomerular amyloid deposition
- tubular disease (because of impaired reabsorption of filtered proteins): chronic interstitial nephritis, polyuric phase of acute tubular necrosis, Fanconi syndrome, tubular toxins (aminoglycosides, lead, cadmium)
- non-renal disease: fever, heavy exercise, heart failure. Orthostatic proteinuria, a benign condition in which proteinuria is present when upright but not when recumbent
- urinary tract disease: infection, tumours, calculi.
- increased production of filterable proteins: immunoglobulin light chains (Bence Jones protein) in myeloma, myoglobinuria, haemoglobinuria
Clinical Presentation
Often asymptomatic (routine screening). Nephrotic syndrome if severe. There may be evidence of underlying cause (e.g. urinary infection, diabetes, hypertension).
Assessment
The history should include enquiries about recent infections, renal disease (including any family history), drugs and occupation. Examination may be normal, but there may be oedema, hypertension, heart failure or evidence of renal failure.
Investigation
- Serum creatinine and electrolytes and ACR or PCR.
- Serum for albumin and protein electrophoresis for monoclonal gammopathy. Blood glucose for diabetes. Urine for free light chains.
- Serum complement (may be low in glomerulonephritis, p. 163), antinuclear antibodies (systemic lupus erythematosis, SLE), antineutrophil cytoplasmic antibodies (systemic vasculitis), cryoglobulin levels.
- Plain abdominal X-ray and ultrasound of renal tract for stones, structural abnormalities and renal size.
In the majority of cases these investigations fail to define the underlying cause, and renal biopsy may be necessary, particularly if nephrotic or there is impaired excretory function. This usually establishes the diagnosis and may identify a treatable cause (particularly some forms of glomerulonephritis).
In the absence of oedema, treatment should be directed towards any underlying cause or associated conditions (e.g. hypertension).
Nephrotic Syndrome
The triad of:
- proteinuria
- hypoalbuminaemia
- oedema.
Aetiology
Any cause of severe proteinuria. Usually it is a consequence of glomerular disease – commonly glomerulonephritis (p. 163), diabetic glomerulosclerosis or renal amyloid. More than 75% of childhood and ~ 20% of adult nephrotic syndrome is a result of minimal-change disease (p. 165). Tubular proteinuria is usually less than 2 g/day and does not cause nephrotic syndrome.
It is associated with thrombosis (loss of anticoagulant proteins such as antithrombin III, protein S, protein C), infection (loss of immunoglobulins) and hyperlipidaemia.
Management
Identify and treat any underlying cause. General management is aimed at the following:
- Reducing oedema with salt restriction and diuretics.
- Angiotensin-converting enzyme inhibitors reduce proteinuria, probably by lowering glomerular capillary pressure. NSAIDs also reduce proteinuria, but these agents reduce renal blood flow and glomerular filtration rate and cause salt retention.
- Treatment of hypertension: angiotensin-converting enzyme inhibitors and diuretics in the first instance, but additional agents may be required.
- Most physicians recommend a normal protein intake.
- Anticoagulate if immobile or thrombotic episode, or severe nephrotic syndrome. Look for and treat intercurrent infection.
- Hyperlipidaemia may be severe. Very-low-density lipoprotein cholesterol, low-density lipoprotein cholesterol and total plasma cholesterol are elevated, as are triglyceride levels. Although this pattern is associated with increased cardiovascular risk, the value of treatment with diet or lipid-lowering agents has not been fully assessed.
Haematuria
Isolated haematuria on dipstick testing of urine can occur in normal individuals. Microscopic haematuria is confirmed by finding more than three red cells per high-power-field of spun urine. Macroscopic haematuria is always abnormal.
Aetiology
Common
- renal tract infection
- renal tract stones (calcium oxalate 80%, triple phosphate 10%, urate 10%, cystine < 1%)
- tumours of the bladder, kidneys and prostate
- glomerulonephritis
- schistosomiasis is common worldwide
Uncommon
- hypertension
- renal trauma
- papillary necrosis
- renal infarction
- drugs – cyclophosphamide (haemorrhagic cystitis), anticoagulants
- medullary sponge kidney (usually benign developmental abnormality with medullary cysts which may be complicated by infection or calculi)
Familial Causes
- polycystic kidneys (p. 159)
- Alport syndrome (p. 159)
- thin basement membrane disease (a generally benign condition in which haematuria is usually the only clinical feature)
- medullary cystic disease (tubulointerstitial nephritis with medullary cysts which usually progresses to renal failure)
The causes vary with age. Glomerular causes predominate in children and young adults, whereas tumours and calculi are common in the elderly.
Investigation
The likely source may be suspected from the history and examination.
Microscopy of a fresh urine sample is performed in all patients to confirm the presence of red cells. The presence of red-cell casts or dysmorphic (abnormally shaped) red cells indicates glomerular bleeding (red cells are deformed by mechanical and osmotic stress as they pass through the tubules). Heavy proteinuria suggests a glomerular lesion, while white-cell casts indicate renal inflammation. Bacteria may be seen and culture should be performed. Urine should also be sent for cytology.
Plasma creatinine to assess renal function.
Plain abdominal film and ultrasound of the renal tract to assess renal size and look for structural lesions (calculi, tumours, cysts).
If glomerular bleeding is suspected (young age, hypertension, proteinuria, renal impairment, absence of structural lesion), consider renal biopsy to identify cause of proteinuria or renal dysfunction.
If a lesion of the renal tract is suspected (older age, no evidence of intrinsic renal disease) proceed to cystoscopy with CT if the upper renal tract has not been clearly identified by ultrasound.
NB Normal urine (centrifuged deposit) contains:
- Red cells 1 × 106 cells/24 h (3 per high-power-field).
- White cells 2 × 106 cells/24 h (6 per high-power-field).
- Hyaline casts are composed of uromucoid (Tamm–Horsfall protein which is excreted by normal tubular cells).
- Cellular casts result from adherence of either red cells (implying glomerular bleeding) or white cells (implying tubular inflammation) to the surface of hyaline casts.
- Epithelial cells may be found in normal urine as a result of contamination by cells from the vulva or prepuce.
Acute Kidney Injury
Characterised by a rapid rise in serum creatinine, usually with a decrease in urine output. The causes can be divided into prerenal, renal and postrenal.
Prerenal
Aetiology
- sepsis – the most common cause, usually complicating surgery or pneumonia
- hypovolaemia from any cause (e.g. haemorrhage, burns, severe diarrhoea or vomiting)
- cardiogenic shock
- drug-induced hypotension (e.g. following drug overdose)
NB Angiotensin-converting enzyme inhibitors may reduce glomerular perfusion sufficiently to cause renal failure if given in the presence of bilateral renal artery stenosis (p. 96). In accelerated (malignant) hypertension, acute, severe hypertension is associated with marked renal abnormalities. The most striking of these is gross intimal hyperplasia, leading to occlusion of the lumen in small arteries and arterioles. Renal failure is a rapid consequence of this condition if the blood pressure is not controlled.
Renal failure commonly complicates advanced liver disease. Plasma urea and creatinine may be normal because of reduced hepatic urea synthesis, low dietary protein intake and loss of muscle mass. There is often a precipitating cause (e.g. hypovolaemia following diuretic therapy, paracentesis or gastrointestinal bleeding, sepsis). Unexplained renal failure complicating liver disease is the hepatorenal syndrome. The prognosis is poor. Reinfusion of ascites into the internal jugular vein via a peritoneo-venous shunt can expand plasma volume and improve renal function, but does not improve survival.
Pathophysiology
Despite high blood flow (20% of cardiac output) the kidneys are particularly susceptible to ischaemia or toxin-induced renal cell injury.
The medulla receives less than 10% of renal blood flow and is at greatest risk of injury. The common response to severe injury (regardless of cause) is acute tubular necrosis (ATN). The necrosis of tubular epithelial cells is most prominent in the proximal tubules and thick ascending limb of the loop of Henle. The tubular lumen may be obstructed by cell debris and casts. Regeneration of tubular cells leading to recovery can take weeks. Severe prolonged ischaemia can cause acute cortical necrosis from which there is little chance of recovery.
The distinction between prerenal failure (in which concentrating powers are retained) and ATN (in which concentrating powers are lost) can be made on urinalysis. In prerenal failure urine osmolality is high (> 500 mosmol/kg), urine sodium is low (< 20 mmol/l) and the urine : plasma urea ratio is > 10 : 1. In ATN urine is isotonic with plasma (< 400 mosmol/kg), urine sodium is > 40 mmol/l and the urine : plasma urea ratio is < 10 : 1.
Renal
Causes
- glomerulonephritis (p. 163)
- nephrotoxic drugs (e.g. aminoglycosides, cyclosporin A, amphotericin B)
- poisoning (e.g. heavy metals)
- myoglobinuria – following rabdomyolysis myoglobin may cause tubular toxicity or form tubular casts. Creatine kinase is markedly elevated
- acute tubular (or cortical) necrosis complicating prerenal disease
- acute interstitial nephritis (usually a drug-induced hypersensitivity reaction which responds to withdrawal of the drug and a short course of corticosteroids. Eosinophils may be present within the predominantly mononuclear cell interstitial infiltrate)
- intrarenal obstruction (e.g. urate or oxalate crystals, calcium precipitation, tubular casts in myeloma)
NB Hypercalcaemia causes renal failure through renal vasoconstriction, direct tubular cell toxicity and distal tubular calcium phosphate precipitation.
Haemolytic–uraemic syndrome (HUS) is characterised by thrombocytopenia (platelet consumption), microangiopathic haemolytic anaemia (red cell fragments on film) and acute renal failure. It commonly follows a diarrhoeal illness in infants infected with a verotoxin-producing strain of Escherichia coli (serotype O157). In adults it may follow an upper respiratory tract infection or be associated with cyclosporin A, the oral contraceptive pill or cytotoxic agents. Familial forms occur due to a mutation in complement factor H. Renal biopsy shows occlusion of glomerular capillaries with fibrin and thrombi, without evidence of complement or immunoglobulin deposition. Recovery usually occurs over a few weeks in children, but the prognosis for adults is poor. Thrombotic thrombocytopenic purpura (TTP, p. 331) is closely related to HUS but is most common in women, and central nervous system involvement and fever are typical additional features. See Trials Box 14.1
 Trials Box 14.1 Management of HUS and TTP
Trials Box 14.1 Management of HUS and TTPPostrenal
Acute urinary tract obstruction from:
- prostatic hypertrophy
- renal and ureteric stones
- tumour of the renal pelvis, ureters or bladder
- blood clot
- sloughed papillae
- external compression from retroperitoneal fibrosis or tumours
- surgical mishap (e.g. ureteric involvement in hysterectomy).
NB Lesions above the bladder must involve both urinary tracts if there are two functioning kidneys.
Investigation
Where there is no obvious cause following careful history and examination, and preliminary biochemical and haematological assessment:
- Check that there is no obstruction. Rectal examination is obligatory to exclude prostatic disease in men, or a pelvic mass. The bladder is enlarged in urethral obstruction. Ultrasound to look for urinary tract dilatation is the simplest method of excluding obstruction, although dilatation may be absent, particularly if obstruction is acute. This will also give information about renal size (small kidneys indicate chronic renal disease; scarring usually indicates chronic interstitial nephritis or ischaemia).
- Proceed to renal biopsy if renal size is normal and there are no clues on investigations, including urinalysis (exclude infection; heavy proteinuria, granular or red cell casts indicate intrinsic renal disease), calcium, uric acid, protein electrophoresis (myeloma), antineutrophil cytoplasm antibodies (vasculitis), antiglomerular basement membrane antibodies (Goodpasture’s disease), antinuclear antibodies (SLE), blood film, platelet, eosinophil count and coagulation (disseminated intravascular coagulation (DIC), TTP, HUS, drug-induced hypersensitivity).
Management
This should be undertaken in a specialised unit where facilities for renal replacement therapy are available.
Identify and correct underlying causes – often multiple (e.g. hypotension, sepsis, DIC and aminoglycoside toxicity).
Rapid correction of prerenal causes (intravenous fluids or blood for hypovolaemia, antibiotics for sepsis, inotropes, avoidance of nephrotoxic drugs) may prevent ATN and restore renal function. Loop diuretics (e.g. furosemide) are often given and may prevent tubular cell ischaemia through inhibition of active sodium chloride reabsorption, thereby reducing oxygen requirements.
Relieve urinary tract obstruction from below (urethral catheterisation with or without ureteric stents) or above (nephrostomy). Prostatic obstruction in elderly men is the most common cause.
Initiate treatment for any intrinsic renal disease (e.g. immunosuppression for certain forms of glomerulonephritis, p. 163).
Continuing assessment of fluid status through input–output records, physical examination, daily weight, lying and standing blood pressure. Fluids should be restricted if there is oliguria or anuria, but patients are usually catabolic and nutrition should not be neglected. A protein intake of 0.6–0.8 g/kg/day with 30 kcal/kg/day should be maintained. In severely ill patients enteral or parenteral nutrition may be necessary.
Careful monitoring of electrolytes, urea, creatinine and acid–base status.
If renal failure persists, renal replacement therapy with haemodialysis or haemofiltration will be required. Absolute indications include hyperkalaemia (potassium above 6–7 mmol/l), markedly elevated plasma creatinine (> 1000 mmol/l, but absolute level must take clinical state into account), severe acidosis (bicarbonate below 10–15 mmol/l) and fluid overload with pulmonary oedema.
Chronic Kidney Disease
Common Causes
- Chronic glomerulonephritis (p. 163).
- Diabetic nephropathy (p. 242).
- Chronic interstitial nephritis (p. 154).
- Hypertension: estimates of the prevalence of chronic renal failure caused by hypertension vary widely, reflecting the fact that the diagnosis of renal disease caused by hypertension depends on the exclusion of other causes. Many cases may have undiagnosed renal disease. Renal failure because of hypertension is much more common in black people than white people, and within the black population there appears to be familial clustering of renal disease caused by hypertension, suggesting a genetic susceptibility to hypertensive renal damage.
- Renovascular disease (p. 96).
- Hereditary renal disease.
- Polycystic kidney disease: an autosomal dominant condition in which there is progressive cystic degeneration of the kidneys. Patients present with hypertension, abdominal pain, haematuria or chronic kidney disease. The diagnosis is confirmed by ultrasound, and family members should be offered screening. Progression to renal failure with hypertension is usual, although the age at which renal replacement therapy becomes necessary varies. Approximately 85% of cases are caused by a defect in PKD1, which maps to 16p13.3, and usually progress more slowly than those due to defects in the PKD2 gene, responsible for most non-16p-linked polycystic kidney disease, and localised to 4q13–q23.
The disease should be considered a multisystem disease in which cysts occur in other organs (liver, pancreas, testes). There is an increased incidence of cardiac valve disease, cerebral aneurysms, hernias and diverticular disease.
- Alport syndrome: 85% of cases are due to mutations in the COL4A5 gene on the X chromosome, which encodes the α5 chain of type IV collagen. Progressive chronic kidney disease associated with sensorineural deafness and eye lesions occurs in affected males, whereas females typically only have abnormalities on urinalysis. Autosomal forms have variable presentations and are due to mutations in the COL4A3 or COL4A4 genes on chromosome 2, encoding the α3 and α4 chains of type IV collagen. Alport syndrome is characterised by thinning and splitting of glomerular basement membrane (GBM). Thin basement membrane disease is a related condition in which thinning of the basement membrane is associated with microscopic haematuria, but renal function is usually preserved. Some patients with thin basemement membrane disease have heterozygous mutations at the COL4A3/COL4A4 locus.
- Long-standing urinary tract obstruction.
NB About 20% of patients with established renal failure present with bilaterally small kidneys and no diagnosis is reached. Under these circumstances renal biopsy is hazardous and unlikely to show reversible changes.
Clinical Features
Screening for renal disease and the availability of dialysis mean that the classical manifestations of uraemia (literally urine in the blood) are now seen infrequently. Chronic kidney disease is typically slow to progress and usually presents with lethargy, general malaise, anorexia and nausea. Generalised pruritus is common. Impotence, menstrual irregularities and loss of fertility are common complaints in younger patients. In severe uraemia there is a characteristic fishy fetor, hiccups, vomiting, severe pruritus with skin excoriations, skin pigmentation, peripheral neuropathy and central nervous system derangements leading to lethargy, stupor and coma with fitting. Pericarditis may be associated with effusion and tamponade.
Investigations
Biochemical
Plasma creatinine provides a guide to the severity of renal failure and can be used to estimate glomerular filtration rate (eGFR). Creatinine is derived from metabolism of creatine in muscle. The rate of production correlates with muscle mass, and depends little on protein intake. Fifty percent loss of renal function is often needed before the serum creatinine rises above the normal range; it is therefore not a sensitive indicator of mild to moderate renal injury.
Urea H2N-CO-NH2 (molecular mass 60 Da) is toxic and is the most abundant nitrogenous compound to accumulate in renal failure. It is the end-product of protein metabolism and is synthesised primarily in the liver. It is freely filtered by the glomerulus, but approximately 50% is reabsorbed so urea clearance is less than glomerular filtration rate (GFR). Urea production increases with cellular catabolism (infection, trauma, steroid therapy) or following protein load (dietary or following gastrointestinal haemorrhage). It is reduced in liver failure.
Renal function is now most commonly reported as estimated GFR (eGFR), which is used to assess the severity of CKD (Table 14.1) and calculated as:
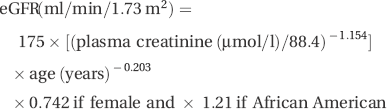
Table 14.1 Classification of Chronic Kidney Disease (CKD) by Severity

This is usually calculated by using a web-based calculator (for example, see http://www.renal.org/eGFRcalc/GFR.pl).
- Cr ethylene diaminetetra-acetic acid (EDTA) clearance more accurately reflects the GFR. It is calculated from the rate of disappearance of a bolus injection of 51Cr EDTA from the blood. The normal GFR is ~100 ml/min per 1.73 m2.
- Hyperkalaemia (p. 169) is common.
- A number of abnormalities of calcium and phosphate homeostasis occur. Phosphate retention is associated with:
 reciprocal depression of serum calcium level
reciprocal depression of serum calcium levelStay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


