Chapter 1 Cell Physiology
Clinical note: There are more than 45 lysosomal storage diseases, caused by impairment of lysosomal function, usually secondary to an inherited deficiency in a hydrolytic enzyme (Table 1-3). The resulting lipid accumulation within lysosomes eventually hinders the activity of cells in many organs, including the liver, heart, and brain. As with I-cell disease, clinical symptoms are severe, and average life expectancy across the entire group of diseases is approximately 15 years, reflecting the importance of normal lysosomal function.
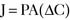
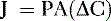
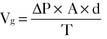
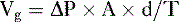
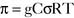
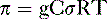
Pharmacology note: Cardiac glycosides such as ouabain and digitalis inhibit the Na+,K+-ATPase (sodium) pump in the myocardium (see Fig. 1-6). This increases the amount of sodium inside the cell, triggering the Ca2+-Na+ countertransporter. More calcium is brought into the cell, which increases the contraction of atrial and ventricular myocardium and increases cardiac output.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


