Name
Amino acids
Topology
Typical paratope
Stability (T m , °C)
Parental
Binders
Knottin
20–50
Knotted 3-disulfide core
Loop 1
~100
N.d.
Affibody
58
Triple α-helical bundle
13 amino acids on surface of helices 1 and 2
72
37–65 (46)
Fibronectin
94
β sandwich
BC + FG (+DE) loops
86
42–73 (57)
DARPin
130–200
4–6 repeats of: β turn + two α helices
Select amino acids in β turn and first α helix
>90
66–85 (79)
Anticalin
150–185
β barrel
12–20 amino acids near “top” of β barrel; dependent on target
79
53–73 (68)
The multitude of scientific, industrial, and in vitro applications empowered by these scaffolds, as well as the optimization of their evolution, has been reviewed [5, 12]. This chapter will provide biophysical information on each scaffold and then focus on the translation of these scaffolds to clinical use in patients; thus, experimental work in animal models and clinical trials are highlighted. Also, at an admitted cost of timeliness, the focus is placed on peer-reviewed published research.
13.2 Fibronectin Domains
The tenth type III domain of human fibronectin (also known as monobody or Adnectin) is a 94-amino-acid (10 kDa) beta sandwich with high thermal stability (T m = 86 °C) (Fig. 13.1) [13]. The scaffold is tolerant to mutations within the loop regions connecting the beta strands, particularly the adjacent loops connecting the BC, DE, and FG strands [14]. Diversification within one to three of these loops has enabled discovery and evolution of specific, high-affinity binding ligands to cell surface receptors, intracellular targets, and extracellular proteins. Affinities typically range from mid-nanomolar to low picomolar [7]. Destabilization due to diversification yields proteins with T m values of 42–73 °C (median 57 °C) [13, 15–17]. It should be noted that incorporation of stability bias in the naïve library elevated the average T m of engineered clones by 10 °C [17]. The scaffold is stable to moderate acidity [18] and devoid of natural cysteines; thus, it is appropriate for use in various intracellular locales. A single thiol can be genetically introduced for site-specific chemistry, or primary amines (N-terminus and lysine at position 63 distal to the paratope) can also be used.
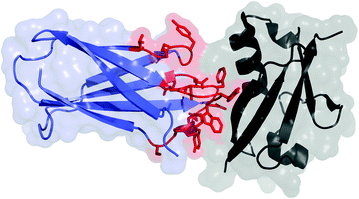
Fig. 13.1
Fibronectin domain (blue and red) in complex with human small ubiquitin-like modifier 1 (black); PDB: 3RZW. The residues commonly mutated in scaffold evolution are shown in red with side chains
13.2.1 CT-322
The fibronectin molecule that has advanced the furthest clinically is CT-322 (BMS-844203, pegdinetanib), a vascular endothelial growth factor receptor 2 binder (VEGFR2) developed by Adnexus Therapeutics (Bristol-Myers Squibb). The lead molecule was discovered by mRNA display [15] and affinity matured for human and mouse cross-reactivity [19]. The evolved protein was modified in several ways as follows: (a) seven amino acids at the N-terminal end, VSDVPRD, were removed; (b) the natural seven-amino-acid linker following the tenth type III domain, EIDKPSQ, was introduced; (c) the serine in the C-terminal tail was mutated to cysteine; and (d) the unique cysteine introduced at the penultimate residue was conjugated to a 40-kDa branched poly (ethylene glycol) (PEG). The resultant molecule has 11 nM affinity for VEGFR2 and negligible cross-reactivity for VEGFR1 or VEGFR3 [20]. PEGylation slows the plasma clearance from a terminal half-time of 4 ± 2 h for native protein to 17 ± 2 h with 20-kDa PEG and 50 ± 20 h with 40-kDa PEG.
In the initial publication of in vivo results for CT-322, the molecule reduced the growth of orthotopic MiaPaCa-xenografted tumors by 42 % in nude mice [21]. Physiological manifestations included increased apoptosis, reduced microvessel density, and reduced VEGF-activated blood vessels. However, metastases were still observed at the same frequency as control treatment. Conversely, in a syngeneic Pan02 xenograft model in immunocompetent mice, CT-322 eliminated metastases although the 23 % reduction in primary tumor size was not statistically significant. Combination therapy with gemcitabine elevated efficacy to a 65 % reduction in tumor size, further validating the therapeutic potential of the evolved scaffold.
Evaluation in a glioblastoma xenograft model in nude mice demonstrated a 45 % reduction in tumor growth [20]. Increasing the dose to 30 mg/kg every other day yielded inhibition of tumor growth and microvessel generation equivalent to anti-VEGFR2 antibody DC101. CT-322 was also compared to small-molecule receptor tyrosine kinase inhibitors. Colo-205 xenograft growth inhibition by CT-322 (30 mg/kg biweekly) was comparable to sorafenib and suritinib but with reduced skin rash, weight loss, and death. Vascular growth was significantly inhibited [22]. CT-322 (60 mg/kg thrice weekly) was comparable to mTor inhibitor temsirolimus, and the benefits were partially additive. Also, in a separate model, the incidence of breast cancer metastasis was inhibited 64 % by CT-322.
Evaluation of CT-322 in an intracranial glioblastoma murine model further validated its efficacy [23]. Monotherapy decreased U87 xenograft tumor size and increased survival from 19 to 29 days. Moreover, CT-322 boosts therapeutic efficacy of the DNA-alkylating agent temozolomide by further decreasing tumor size and extending survival from 32 to 47 days. Immunohistochemical staining demonstrates inhibited microvessel formation, increased apoptosis, and decreased proliferation.
Therapeutic efficacy in multiple murine models with concomitant validation of physiological mechanisms motivated further clinical translation of CT-322. Toxicology studies indicated no adverse effects at 5 and 10 mg/kg in rats and monkeys [24], further supporting human trials. A phase I trial identified a maximum tolerated dose of 2 mg/kg intravenously weekly or biweekly, which yielded a lack of severe toxicity and a clearance half-time of 71–98 h [24]. 82 % of patients developed anti-drug antibodies that were shown to bind to the engineered loops of CT-322, which are of synthetic, non-human origin. Nevertheless, the response did not impact CT-322 or VEGF-A concentrations. Of the 34 patients available for therapeutic evaluation, 68 % had stable disease and 12 % demonstrated a reduction in tumor size.
In a phase II study for first-line treatment of advanced non-small-cell lung cancer, patients received paclitaxel/carboplatin plus either CT-322 or bevacizumab (antibody targeting VEGF-A) [25]. Progression-free survival (5.6 months vs. 6.8 months; CT-322 vs. bevacizumab), overall survival (12.5 months vs. 15.2 months), and response rate (25 % vs. 33 %) were all lower with CT-322 than with bevacizumab. Thrombocytopenia and grade ≥3 hypertension increased with CT-322. The efficacy of CT-322 in other clinical indications awaits further study.
13.2.2 Additional Therapeutic Fibronectins
An additional growth factor receptor antagonist, a type I insulin-like growth factor receptor (IGF1R) binder with 110 pM affinity, has been studied [26]. This molecule, PEGylated akin to CT-322, reduced vascular surface area by 59 % and reduced tumor growth by 37 % in a xenograft mouse model of Ewing’s sarcoma. Combination treatment with CT-322 did not yield statistically significant gains in tumor inhibition.
This agent was also genetically fused to an epidermal growth factor receptor (EGFR)-targeted fibronectin [27]. PEGylation reduced the affinity of both components 10–20 fold though strong parental affinity yielded 1 and 10 nM final affinity for IGF1R and EGFR. The bispecific agent inhibits H292 lung cancer xenografted tumors in mice compared to EGFR-targeted panitumumab. More notably, the bispecific fibronectin construct inhibits RH41 pediatric rhabdomyosarcoma xenografted tumors, which are not effectively treated by panitumumab. In BxPC3 xenografted tumors, the bispecific yielded greater tumor growth inhibition than EGFR-targeted cetuximab or even a mixture of monospecific fibronectin domains. Thus, bispecific agents may provide benefits over multi-agent treatment approaches. Notably, the fibronectin-based bispecific agent was a simple genetic fusion with a (GS)10 linker and was stable (T m = 58 °C) and monomeric. 14C labeling of the domain along with whole-body autoradiography delineated physiological distribution including variable tumor uptake (1–3 %ID/g in RH41 vs. 6–11 %ID/g in H292) [28].
Research on an alternative multi-specific agent provides data on one possible mechanism for improved efficacy relative to monospecific agents. An antibody–fibronectin construct targeting three independent EGFR epitopes clusters receptors, yielding downregulation of surface EGFR. This construct dramatically reduced the growth of BRAF and KRAS xenografted tumors in mice [29].
13.2.3 Diagnostics
On the diagnostic front, the fibronectin domain has demonstrated effective imaging in molecular positron emission tomography (PET). A fibronectin domain engineered for binding EGFR was radiolabeled with 64Cu using a 1,4,7,10-tetra-azacyclododecane-1,4,7,10-tetraacetic acid (DOTA) chelator [30]. The resultant molecule is stable in mouse and human serum for at least 24 h. Molecular PET exhibits EGFR-specific tumor uptake within ~20 min and rapid clearance from blood (half-time = 2.1 min) and healthy tissue. 3.4 ± 1.0 %ID/g was achieved in tumor at 1 h post-injection with a 9:1 tumor:background ratio. The primary limitation of the molecule is high renal uptake and retention although the 2.1 mGy/MBq renal dose is still only 2 % of the maximum tolerated dose. Moreover, preliminary work indicates that using an 18F-labeling approach substantially reduces renal signal. The higher positron yield and more rapid radioactive decay (half-life = 1.8 h) make 18F a preferable isotope for clinical use. The impact of protein scaffold hydrophilicity and charge on physiological distribution was elucidated using molecular PET to analyze delivery of fibronectin mutants [31]. Renal retention was reduced by selective removal of charged residues. Hydrophilicity reduces hepatic retention but elevates renal retention.
13.2.4 Future Outlook for Fibronectin
Fibronectin domains have been effectively used as monovalent antagonists, multi-specific fusions, immunotoxins in cell culture [32], and molecular PET diagnostics. A PEGylated fibronectin has proven safe and biologically active in patients. Additional study will continue to identify the efficacy of this compound and others relative to alternative therapeutic molecules in various indications including cardiovascular disease [33]. The small size and rapid clearance of non-PEGylated fibronectins are well suited for molecular imaging, which has demonstrated preclinical efficacy. Efficacy in both therapy and imaging coupled with robust, efficient evolution of stable, high affinity binders to a multitude of targets poise the fibronectin scaffold for continued successful development.
13.3 Knottins
Knottins are polypeptides, typically 25–50 amino acids in length, with at least three disulfide bonds that result in a knotted structure (Fig. 13.2). An additional subset of knottins, called cyclotides, have connected N- and C-termini to yield cyclic knots. The knotted core provides exceptional stability, including the ability to withstand near-boiling temperatures and denaturants [34]. Knottins have demonstrated stability to gastric proteases and provide potential for oral delivery [35]. The small size is well suited to rapid, efficient physiological distribution. The molecule can be prepared recombinantly, typically in the methylotropic yeast Pichia pastoris [36], or synthetically, in which the complex knotted structure requires careful refolding. The knotted structure yields several solvent-accessible loops between cysteine residues; these loops exhibit sequence and length variability across the multitude of knottins [37]. Appropriate sequences within one or two of these loops can provide binding to a host of targets.
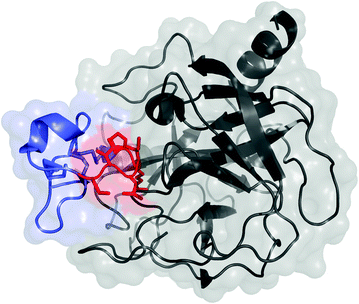
Fig. 13.2
Knottin peptide (blue and red) in complex with trypsin (black); PDB: 1H9I. The residues commonly mutated in scaffold evolution of loop 1 are shown in red with side chains
13.3.1 Natural Knottins
Several natural knottins have been explored for clinical use. For example, ziconotide, isolated from the cone snail, is an N-type calcium channel antagonist. It is approved by the United States Food and Drug Administration (FDA) as an analgesic for severe chronic pain. It has also demonstrated 44 % reduction in ischemic cerebral damage in a rabbit model [38]. In mouse models of cachexia, a 50-amino-acid portion of agouti-related peptide (AgRP) targeting melanocortin receptor has effectively inhibited appetite suppression and weight loss induced by radiation or tumor growth [39]. The 36-amino-acid chlorotoxin targets annexin A2, resulting in preferential binding to multiple malignant cell types relative to healthy tissue. Conjugation with 131I enables single-photon emission computed tomography (SPECT) imaging of glioma in human patients [40] and has completed phase I and II trials for radiotherapy [41, 42]. Conjugation of the near-infrared fluorophore Cy5.5 has enabled fluorescence molecular imaging of xenografted glioma, spontaneous medulloblastoma, primary prostate tumors, and lung metastases in mice [43]. Decoration of superparamagnetic iron oxide nanoparticles with chlorotoxin permits molecular magnetic resonance imaging [44].
13.3.2 Synthetic Knottins
Synthetic knottins have been engineered by grafting binding motifs into knottin loops and evolving the grafted peptides. Thrombopoietin peptide mimics were grafted into knottin frameworks based on AgRP or Ecbalium elaterium trypsin inhibitor II (EETI-II) and crosslinked [45]. The resultant dimers stimulated proliferation of megakaryoblasts. A 200 ng dose of an EETI-II-based dimer was able to double the platelet count in a mouse model.
The Cochran lab in conjunction with the Cheng and Gambhir labs has performed extensive murine studies on knottins for molecular imaging of integrins. The RGD tripeptide motif was inserted into loop 1 of the EETI-II scaffold, and directed evolution with yeast surface display was used to identify clones with ~20 nM affinity for integrin αvβ3 [46]. These clones were then used for development of molecular imaging agents. Conjugation with Cy5.5 or 64Cu enabled near-infrared fluorescence imaging or PET of xenografted U87 tumors in nude mice [47]. At 4 h post-injection, 4 %ID/g tumor and a 20:1 tumor:muscle ratio were achieved for the radiolabeled agent. Simultaneous labeling with the fluorophore and radioisotope was also achieved while retaining activity [48]. To better match the rapid biodistribution and clearance of the 4-kDa knottin, clone 2.5D was radiolabeled with 18F using N-succinimidyl-4-fluorobenzoate chemistry [49]. Effective tumor imaging was retained although tumor uptake (2.6 ± 0.7 %ID/g at 0.5 h, 1.5 ± 0.4 %ID/g at 1 h), and specificity (5:1 tumor:muscle at 0.5 h, 3:1 tumor:muscle at 1 h) reduced slightly. 64Cu-labeled clone 2.5F exhibited superior specificity relative to 18F-fluorodeoxyglucose for imaging nascent lung nodules in a transgenic mouse model (6.0 ± 0.6 tumor:background for 64Cu-knottin vs. 4.4 ± 0.7 for 18F-fluorodeoxyglucose) [50]. The 2.5D clone was also used for molecular ultrasound [51]. Conjugation to perfluorocarbon-filled microbubbles yielded specific detection of integrin αvβ3-expressing tumors in murine models. Targeting was superior to microbubbles labeled with integrin αvβ3 antibodies or cyclic RGD peptides and was molecularly specific as verified by blocking and cellular control studies.
The diversity of the knottin family was also demonstrated as the AgRP scaffold was used to engineer nanomolar affinity binders to integrin αvβ3 [52] and label them with 64Cu for PET [53]. Imaging of xenografted U87 tumors in nude mice was effective although tumor uptake was slightly lower (2.7 ± 1.1 %ID/g at 1 h vs. 4.6 ± 0.6 %ID/g) and renal signal much higher (60 ± 18 %ID/g vs. 4 ± 1 %ID/g) than with EETI-II. 111In labeling reduced renal signal to 34 ± 8 %ID/g but hindered tumor retention [54]. 18F labeling using 4-nitrophenyl-2-fluoropropionate reduced renal signal to 20 %ID/g [55]. Evolution of an additional scaffold, the Momordica cochinchinensis trypsin inhibitor-II (MCoTI-II), further substantiated the breadth of the knottin scaffold, including application to a new integrin target, αvβ6 [56]. Clones with low nanomolar affinity were evolved by loop grafting and directed evolution with yeast surface display. Ligands were labeled with 64Cu for molecular PET of nude mice with subcutaneous or orthotopic xenografted pancreatic cancers. The original, arginine-rich scaffold yielded 5 ± 1 %ID/g tumor with an 8 ± 2 tumor:background ratio at 1 h post-injection albeit with 75 ± 5 %ID/g kidney. Transfer of the evolved loop sequence to a serine-rich scaffold decreased renal uptake to 18 ± 3 %ID/g but also decreased tumor uptake to 1.8 ± 0.4 %ID/g. Renal retention was substantially reduced with 18F radiolabeling using N-succinimidyl-4-fluorobenzoate chemistry (9 ± 3 %ID/g kidney) albeit with slight tumor reduction (2.3 ± 0.6 %ID/g) [57].
Radiolabeled knottins were also evaluated for radiotherapy. 177Lu-labeled integrin αvβ3 binders 2.5D and 2.5F were administered to mice bearing U87 tumor xenografts [58]. 61 mGy/mCi absorbed dose was achieved in the tumor with 18 mGy/mCi in the kidneys and <2 mGy/mCi elsewhere.
13.3.3 Future Outlook for Knottins
Natural knottins have demonstrated efficacy in a range of applications but are limited to targeting their native binding partners. Loop grafting and evolution in multiple knottin frameworks have proven highly efficacious for integrin targeting in a variety of murine models, primarily across multiple modalities of molecular imaging but also for radiotherapy. The breadth of impact for the knottin family will depend on the ability to robustly and efficiently develop high-affinity binders to a multitude of targets. To aid in affinity, an integrin binding motif was simultaneously grafted into two loops of the EETI-II scaffold, which enhanced binding and maintained effective molecular PET [59]. Looking to expand the evolutionary capacity of the scaffolds beyond integrins, the EETI-II scaffold has been evolved for binding to seven different targets albeit with limited affinity evaluation [60]. If high affinity can be readily achieved from naïve libraries without grafted motifs, the stable, small knottin family of scaffolds has tremendous potential for molecularly targeted imaging and therapy.
13.4 DARPins
DARPins comprise ankyrin repeat domains, typically four to six repeats each of which has a beta turn and two antiparallel alpha helices [8] (Fig. 13.3). A tight hydrophobic core, improved by consensus design, imparts tremendous stability. This is evidenced by robustness to boiling and guanidine hydrochloride and tremendously slow amide proton exchange. While the sequence is fully non-human, immunogenicity is expected to be limited as the inherent stability limits degradation. Molecular size (~15 kDa depending on the number of repeats) provides reasonable permeability and diffusion along with rapid plasma clearance. DARPins are very well expressed in the soluble phase of bacterial culture (≥100 mg/L in shake flask and ≥1 g/L in a fermenter). Diversification in the beta turn and the first alpha helix presents a rigid potential paratope with extensive surface area. Ligands with nanomolar to picomolar affinities for many targets have been evolved.
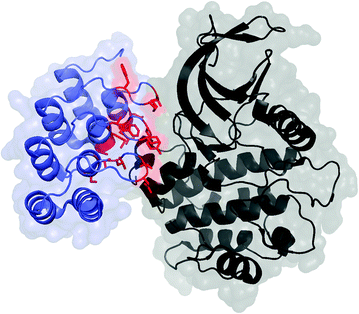
Fig. 13.3
DARPin (blue and red) in complex with polo-like kinase 1 domain (black); PDB: 2V5Q. The residues commonly mutated in scaffold evolution are shown in red with side chains. C-terminal capping domain has been omitted
13.4.1 Applications of DARPins
DARPins have demonstrated efficacy in a variety of applications. A DARPin was evolved by ribosome display to achieve 90 pM affinity for human epidermal growth factor receptor 2 (HER2) [61]. The ligand was labeled with 99mTc and yielded effective localization of a murine tumor xenograft via SPECT/CT [62]. Biodistribution quantitation indicated 9 ± 2 %ID/g tumor with 13 ± 3 tumor:blood at 1 h; 8 ± 3 %ID/g tumor, 27 ± 15 tumor:blood at 4 h; and 8 ± 2 %ID/g tumor, 62 ± 28 tumor:blood at 24 h. Kidney and liver backgrounds were 239 ± 33 %ID/g and 7.3 ± 1.0 %ID/g, respectively, at 4 h. Extensive characterization was performed to quantify the impacts of affinity and size. While protein dimerization had a nearly negligible impact on plasma clearance (half-time of ~3 min), PEGylation drastically slowed clearance (half-time = 19 h for 20 kDa PEG; 50 h for 40 and 60 kDa PEG). This pharmacokinetic modulation decreased tumor:blood ratio from 13 ± 3, 27 ± 15, and 72 ± 27 tumor:blood at 1, 4, and 24 h post-injection for native protein to 0.2 ± 0.1, 0.6 ± 0.3, and 4.7 ± 3.1 for the PEGylated protein. Tumor uptake correlated with affinity for (a) the 14.5 kDa native proteins from 1 to 48 h and (b) the PEGylated constructs at later time points.
The 90 pM HER2 binder was also efficacious for in vitro diagnostics. The DARPin exhibited superior sensitivity and specificity relative to a validated antibody in immunohistochemistry of breast cancer tissue microarrays [63]. Picomolar affinity was critical to success as a DARPin mutant with 1 nM affinity was significantly less effective.
HER2 DARPins were also effective in viral targeting. Fusion of a hemagglutinin mutant with an anti-HER2 DARPin enabled molecular targeting of lentiviral vectors [64]. Vector particles were systemically administered into mice with subcutaneous tumor xenografts of HER2-positive (SKOV3) or HER2-negative (U87MG) tumors. Using a luciferase reporter, HER2-specific viral targeting was evident within 3 days and was retained for at least 14 days. Oncolytic recombinant measles virus was also effectively targeted with an anti-HER2 DARPin [65]. Tumor inhibition following intratumoral injection of a DARPin-targeted virus was superior to an antibody-targeted virus; inhibition was comparable to untargeted virus, but without associated toxic side effects. Also, a dual-targeted (HER2 and epithelial cell adhesion molecule (EpCAM)) bispecific virus demonstrated improved resistance to tumor modulation in cell culture.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


