Osteoarthritis (OA)
This common degenerative disorder, characterised by inadequate repair of cartilage and periarticular bone in response to damage or injury, ultimately leads to joint failure. Radiological evidence of OA can be detected in a quarter of the population by their mid-40s, and in virtually everyone by their mid-60s. Both sexes are affected, although severe disease and hand involvement are seen more frequently in women. The most common joints to be affected are the interphalangeal joints, the 1st carpometacarpal joint, cervical and lumbar spine, knees and hips.
Genetic factors play a role in the pathogenesis of OA but remain poorly understood. Obesity increases the prevalence of OA in the weight-bearing joints of the lower limb. Other predisposing factors include trauma, meniscectomy, joint inflammation, neuropathic joints, acromegaly, haemochromatosis, haemoglobinopathies, alkaptonuria and Gaucher’s disease.
OA is characterised by progressive disruption and loss of hyaline cartilage, with sclerosis, cysts and osteophyte formation in underlying subchondral bone and narrowing of the joint space. Secondary changes are seen in the adjacent synovium.
Clinical Presentation
OA may be asymptomatic (especially in the spine). Clinical features vary depending on the joint(s) involved, but symptoms include the following:
- joint pain, worse with movement and towards the end of the day
- stiffness
- swelling, e.g. of the distal and proximal interphalangeal joints with Heberden’s and Bouchard’s nodes respectively.
On examination there is tenderness, bony swelling (osteophyte formation), painful restriction of movement, crepitus and, if long-standing, muscle wasting. Joints may be red, warm and tender and associated with an effusion (synovial inflammation).
Functional impairment, immobility, deformity and occasionally nerve (e.g. carpal tunnel syndrome) or nerve root (cervical or lumbar spine) entrapment may all complicate OA.
Investigation
Diagnosis depends on clinical assessment and radiological findings. Plain radiographs typically reveal:
- loss of joint space (cartilage loss)
- osteophytes
- sclerosis of subchondral bone
- ± bone cyst formation.
Management
- Analgesia – initially paracetamol and NSAIDs.
- Weight loss in obese subjects.
- Physiotherapy and graded exercise help to maintain muscle bulk and strength.
- Walking aids and orthotics may offer effective symptomatic relief.
- Intra-articular corticosteroids where there is worsening pain and evidence of synovial inflammation (warmth, effusion).
- Joint replacement especially in those with reduced mobility and rest or nocturnal pain.
- Domestic and mobility aids.
Prognosis
Usually the condition slowly progresses over time, although in some patients symptoms will abate.
Rheumatoid Arthritis (RA)
The commonest cause of polyarticular joint inflammation, RA is characterised by a distinct pattern of joint involvement often accompanied by extra-articular disease manifestations. It occurs throughout the world, with an estimated prevalence of 1%. Women are more frequently affected than men ( 3 : 1), with a peak age of onset between 40 and 60 years, although RA may present as early as young adulthood.
3 : 1), with a peak age of onset between 40 and 60 years, although RA may present as early as young adulthood.
RA is associated with certain HLA haplotypes, including HLA-DR4. HLA-DR4 positivity is associated with erosive seropositive disease. Environmental factors that have been implicated in the development of RA include cigarette smoking, diet, hormonal changes and infections, although epidemiological studies have failed to establish a causal link with any specific organism.
Synovial inflammation is the hallmark of RA. In the early stages there is disruption of the synovial microvasculature, followed by synovial thickening and heavy infiltration with lymphocytes, macrophages and plasma cells. The latter may secrete rheumatoid factors (RF; see below). Inflamed hypertrophied synovium (pannus) encroaches on the adjacent cartilaginous surface, resulting in thinning of the cartilage and erosion of the underlying bone. An array of cytokines (interleukin-1 (IL-1), IL-2, IL-4, IL-6 and tumour necrosis factor-α (TNF-α)) have been implicated in the pathogenesis of RA.
Clinical Presentation
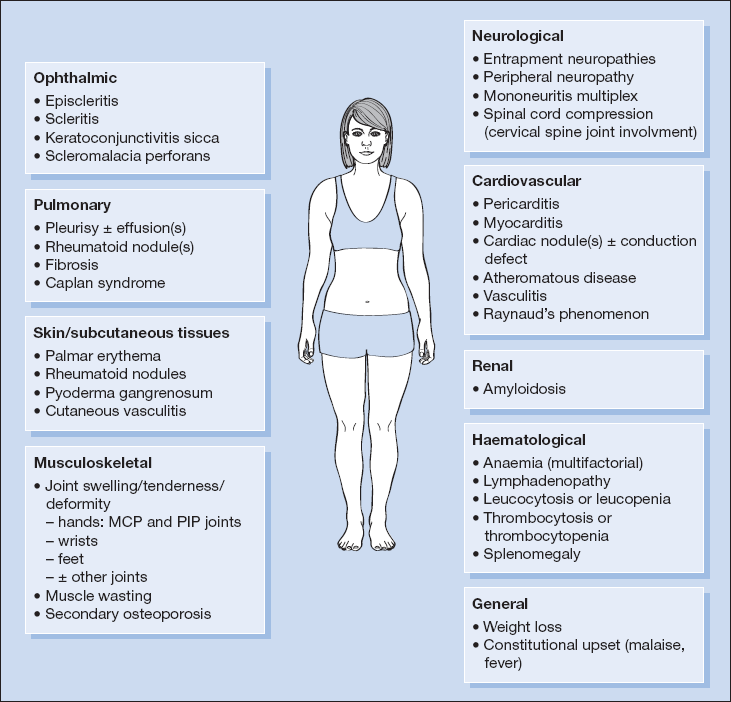
RA is a multisystem disorder in which extra-articular manifestations are key to long-term outcomes. Extra-articular disease is more likely in RF seropositive patients. Figure 18.1 summarises the diverse array of clinical features associated with RA.
Musculoskeletal System
The small joints of the hands and feet are the most commonly affected, usually symmetrically, but large synovial joints (hips, knees, elbows) are often involved. Gradual onset with progressive pain, early-morning stiffness and swelling of joints is usual, although acute onset associated with fever and general malaise is recognised.
Hands
- symmetrical polyarthropathy involving proximal joints
- tenderness and diminished movement of involved joints:
 sparing of the terminal interphalangeal joints
sparing of the terminal interphalangeal joints the wrists are commonly involved
the wrists are commonly involved- deformity due to joint subluxation and tendon misalignment
 swan-neck
swan-neck boutonnière
boutonnière z-deformity of the thumb
z-deformity of the thumb metacarpophalangeal subluxation
metacarpophalangeal subluxation ulnar deviation at the metacarpophalangeal joints
ulnar deviation at the metacarpophalangeal joints- swelling:
 fusiform soft-tissue swelling of the metacarpophalangeal (MCP) and proximal interphalangeal (PIP) joints
fusiform soft-tissue swelling of the metacarpophalangeal (MCP) and proximal interphalangeal (PIP) joints soft tissue involvement causes swelling, tenosynovitis and tendon rupture
soft tissue involvement causes swelling, tenosynovitis and tendon rupture- wasting:
 combination of disuse atrophy, vasculitis and peripheral neuropathy
combination of disuse atrophy, vasculitis and peripheral neuropathy- reduced function
Other Joints
- Elbows, shoulders and knees commonly involved.
- Ankles, costovertebral, temporomandibular and the cricoarytenoid joints may be affected.
- Cervical spine: the axial skeleton is generally spared, except for the cervical spine – laxity of the atlantoaxial joint ligaments with erosion of the odontoid peg may result in acute or chronic cord compression. Subluxation is life-threatening and should be particularly considered if general anaesthesia is required.
Skin and Subcutaneous Tissues
- palmar erythema
- rheumatoid nodules – typically on the ulnar border of the forearms and only in patients who are rheumatoid factor positive
- scars of previous surgery (e.g. metacarpophalangeal joint replacement, excision of the ulnar styloid process, extensor tendon repair, carpal tunnel release)
- pyoderma gangrenosum
- vasculitis including lower limb ulceration
- thin, fragile skin in those on long-term corticosteroid therapy with other features of iatrogenic Cushing syndrome)
Ocular
- episcleritis/scleritis
- keratoconjunctivitis sicca in approximately 15% of patients with RA
- scleromalacia perforans
- iatrogenic – lens opacities and retinal degeneration with chloroquine treatment and cataracts from corticosteroid therapy
Pulmonary
- Clinical signs of pulmonary involvement uncommon.
- Lung function tests show changes in up to 50% of all patients with seropositive RA.
Presentations include:
- pleural effusion(s)
- rheumatoid nodules – single or multiple
- diffuse fibronodular infiltration or fibrosis
- Caplan syndrome – the presence of multiple round well-defined nodules (typically 0.5–2 cm, but in some cases as large as 5 cm in diameter) in the lungs of miners (coal, silicosis, asbestos) with RA; they may calcify, cavitate or coalesce and be mistaken for tuberculosis.
Cardiovascular
- pericarditis (with or without effusion)
- myocarditis
- cardiac nodules
Vascular manifestations include:
- arteritic lesions:
 nail fold infarcts
nail fold infarcts ‘splinter’ necrosis in the digital pulps
‘splinter’ necrosis in the digital pulps necrotising arteritis affecting larger vessels causing digital gangrene, bowel infarction or stroke
necrotising arteritis affecting larger vessels causing digital gangrene, bowel infarction or stroke vasculitis
vasculitis Raynaud’s phenomenon.
Raynaud’s phenomenon.Neurological
- entrapment neuropathies (e.g. carpal tunnel syndrome, ulnar neuropathy)
- peripheral neuropathy – predominantly sensory, secondary to arteritis or complicating drug therapy
- mononeuritis multiplex – usually digital, ulnar and lateral popliteal nerves
- spinal cord compression – secondary to cervical spine joint involvement
Haematological (Table 18.1)
- Normochromic normocytic anaemia is common and its severity relates to that of the underlying disease.
- Iron deficiency secondary to drug therapy.
- The height of the ESR reflects the activity of the disease.
- CRP is raised.
- Leucocytosis may signify active disease, intercurrent infection or corticosteroid therapy.
Table 18.1 Causes of anaemia in RA
| Type | Cause |
| Normochromic normocytic | Anaemia of chronic disease |
| Hypochromic microcytic | Iron deficiency secondary to aspirin and other NSAIDs (e.g. ibuprofen) |
| Macrocytic | Folate deficiency secondary to methotrexate or sulphasalazine; vitamin B12 deficiency (due to associated pernicious anaemia) |
| Haemolytic | Drug-induced (e.g. sulphasalazine, dapsone) |
| Bone marrow suppression | Drug-induced (e.g. sulphasalazine, gold, cytotoxics) |
| Hypersplenism | Felty syndrome |
Reticuloendothelial System
- Generalised lymphadenopathy is present in up to 10% of cases.
- The spleen is enlarged in about 5% of patients and 1% develop leucopenia.
- Felty syndrome: the triad of RA, splenomegaly with neutropenia and thrombocytopenia, and lymphadenopathy may also be present.
Renal
- Amyloidosis is now less common in RA, reflecting improved disease control.
- Proteinuria or overt nephrotic syndrome may complicate treatment with pencillamine and gold.
Iatrogenic
Clinical evidence of side effects of therapy may be observed (see below).
The American College of Rheumatology has proposed criteria (1987) for the diagnosis of RA based on these clinical features (Box 18.1).
 Box 18.1 American College of Rheumatology (ACR) Revised Criteria for the Diagnosis of RA
Box 18.1 American College of Rheumatology (ACR) Revised Criteria for the Diagnosis of RA| To establish a diagnosis of RA, ≥ four of the following criteria are required: | ||||
| •morning stiffness of > 1 h duration most mornings for > 6 weeks | ||||
| •arthritis involving at least three areas (soft tissue swelling or fluid) for > 6 weeks | ||||
| •arthritis of hand joints for > 6 weeks | ||||
| •symmetrical arthritis for > 6 weeks | ||||
| •rheumatoid nodules | ||||
| •positive rheumatoid factor | ||||
| •radiological changes of RA (wrists, hands) | ||||
| NB These criteria were originally intended for research categorisation; however, in practice it would be unwise to defer treatment until a patient meets all of the ACR criteria for RA as early intervention is paramount for preserving long-term structure/function. | ||||
Investigation
Currently there are no laboratory or radiological tests that reliably confirm or rule out RA in all patients and clinical impression remains crucial to diagnosis. A high index of suspicion and early onward referral for expert opinion are recommended as uncontrolled inflammation translates into joint damage and subsequent disability.
Serology
Rheumatoid Factor (RF)
High titres of IgM RF correlate with more severe arthritis and with extra-articular disease. RF is not specific to RA, being found in low titres in  5% of the general population (and does not predict disease in clinically normal individuals), in Sjögren syndrome and in other connective tissue disorders. Some patients with RA remain seronegative.
5% of the general population (and does not predict disease in clinically normal individuals), in Sjögren syndrome and in other connective tissue disorders. Some patients with RA remain seronegative.
Recently, alternative serological tests appear to offer comparable sensitivity but improved specificity to RF for diagnosing RA, e.g. anti-citrullinated protein antibodies (ACPAs), including anti-CCP (cyclic citrullinated peptide).
Radiology
The joints may be radiologically normal in the earliest disease stages. The characteristic sequence of abnormalities is:
- soft-tissue swelling and periarticular osteoporosis
- narrowing of joint space and periarticular erosions
- subluxation and osteoarthritis (in long-standing disease); and finally
- fibrosis or bony ankylosis.
Management
Assessment of disease activity depends on both clinical and laboratory findings. The objectives of therapy are:
- symptom relief – in particular control of pain and stiffness
- suppression of active disease and arrest of disease progression
- restoration of joint function.
This requires a multidisciplinary team (MDT) approach involving rheumatologists, physiotherapists, occupational therapists, orthopaedic surgeons, specialist nurses and social services. Patient education should involve information about the disease chronicity and tendency to cycle between exacerbations and remissions. Patients should have a named contact (usually a specialist nurse) who can ensure rapid access to the team in the event of a disease flare. The UK National Institute for Health and Clinical Excellence (NICE) issued guidelines in 2009 emphasising the importance of the MDT in ensuring high quality care for patients with RA (Box 18.2).
 Box 18.2 Summary of National Institute for Health and Clinical Excellence (NICE) 2009 Guidance for the Management of Rheumatoid Arthritis in Adults*
Box 18.2 Summary of National Institute for Health and Clinical Excellence (NICE) 2009 Guidance for the Management of Rheumatoid Arthritis in Adults*| •Referral, diagnosis and investigations – consider early serological and radiological screening and referral for expert review in all suspected cases |
| •Communication and education – offer verbal and written information to patients with RA; encourage involvement in self-management programmes |
| •MDT – ensure ongoing regular access to the individual members of the MDT (e.g. physiotherapist, occupational therapist, podiatrist); patients should have easy access to a named point-of-contact (e.g. nurse specialist) |
| •Management of symptoms: analgesics and NSAIDS – offer simple analgesics if pain control is inadequate; NSAIDs and COX-2 inhibitors should be used at the lowest effective dose for the shortest time possible (choice of agent should be decided on an individual basis), with coprescription of a PPI to provide gastric protection; if symptom control is inadequate, review DMARDs/‘biologics’ regimens |
| •Management of symptoms: DMARDs: |
| •For newly diagnosed active disease – offer a combination of DMARDs; ideally include methotrexate + at least one other agent + short-term corticosteroids |
| •For recent onset disease (< 2 years) – once sustained and satisfactory disease control established, cautiously try to reduce dosages of DMARDs |
| •For established disease (> 2 years) – if disease is stable, cautiously reduce dosages of DMARDs or ‘biologics’, but return promptly to disease-controlling regimens at the first sign of a flare-up; when introducing new drugs to improve disease control, consider decreasing or discontinuing pre-existing agents |
| •Management of symptoms: corticosteroids: |
| •For recent onset or established disease – offer short-term courses for flare-ups |
| •For established disease – continue long-term therapy only after careful discussion with the patient regarding adverse effects, and after offering all other treatment options |
| •Monitoring RA – regularly monitor CRP and key components of disease activity (e.g. using a composite score such as DAS28 that includes assessment of 28 joints) to help guide treatment decisions; arrange regular clinic/specialist nurse follow-up; check for comorbidities (e.g. hypertension, ischaemic heart disease, osteoporosis, depression); assess for complications (e.g. ocular involvement, disease of the cervical spine) |
| •Timing and referral for surgery – offer early referral for specialist surgical opinion when there is persistent pain, worsening joint deformity/function or persistent synovitis despite medical therapy; urgent surgical review is also indicated when there is imminent/actual tendon rupture, nerve entrapment, stress fracture or evidence of cervical myelopathy |
| •Diet and complementary therapies – for patients wishing to experiment with their diet explain that currently there is no strong evidence that their arthritis will benefit; advice to follow a ‘Mediterranean diet’ is reasonable; advise that there is little or no evidence for complementary therapies offering long-term efficacy in RA, and therefore if tried these should not replace conventional treatment even if they yield short-term symptomatic benefit |
| *National Institute for Health and Clinical Excellence (2009) The Management of Rheumatoid Arthritis in Adults. Clinical Guideline 79. NICE, London. www.nice.org.uk/CG79; reviewed in Deighton et al., BMJ 2009; 338: 710–712. |
During the active phase, treatment involves both local measures (physiotherapy, use of splints, intra-articular corticosteroids) and systemic drug therapy.
Drug therapy
- Simple analgesics: help some patients with mild disease.
- NSAIDs: useful symptom relief but do not alter the underlying disease process. Should not be used in isolation and long-term use is limited by side effects.
- Cyclo-oxygenase-2 inhibitors: an alternative to NSAIDs, contraindicated in patients at risk of vascular disease.
Disease-Modifying AntiRheumatic Drugs (DMARDs)
A heterogenous group of drugs for use under expert supervision. Treatment should be commenced once the diagnosis has been established, and not delayed until complications develop. Combination therapy is usually favoured:
- Methotrexate: first-line DMARD provided there are no contraindications to its use. Given weekly with folic acid supplementation on a different day; adverse effects include gastrointestinal disturbance, bone marrow suppression, hepatotoxicity, pneumonitis, renal damage.
- Sulphasalazine: adverse effects include gastrointestinal upset, skin rashes, bone marrow suppression, hepatotoxicity.
- Hydroxychloroquine (chloroquine): adverse effects include ocular toxicity, particularly with chloroquine, gastrointestinal upset, skin reactions, seizures, myopathy and psychiatric disturbance.
- Azathioprine: adverse effects include gastrointestinal upset and bone marrow suppression.
- Leflunomide: adverse effects include gastrointestinal upset, raised blood pressure, bone marrow suppression, hepatotoxicity.
- Gold (intramuscular sodium aurothiomalate or oral auranofin): adverse effects include oral ulceration/stomatitis, irreversible skin pigmentation in sun-exposed areas, gastrointestinal upset, hepatotoxicity, blood dyscrasias (may be sudden and fatal), nephrotic syndrome.
- Penicillamine: adverse effects include gastrointestinal upset, transient loss of taste, skin disorders, bone marrow suppression, cholestatic jaundice.
- Ciclosporin and cyclophosphamide may be effective in severe disease refractory to other agents.
Tumour Necrosis Factor-α Inhibitors (Anti-TNFα Therapies)
These are generally reserved for use in patients who fail conventional DMARD therapy. Three drugs are currently available: infliximab and adalimumab are monoclonal anti-TNFα antibodies, and etanercept is a soluble TNF receptor. All are given by injection and methotrexate should be continued if possible. Adverse side effects include hypersensitivity reactions/anaphylaxis, gastrointestinal upset, increased susceptibility to infections including tuberculosis and hepatitis B reactivation, bone marrow suppression and cardiac failure.
Rituximab
Rituximab is reserved for the treatment of severe active RA in patients whose condition has not responded adequately to DMARDs and at least one TNFα inhibitor. It works by depleting circulating B-cells and is used in conjunction with methotrexate. It predisposes to infection.
Corticosteroids
Glucocortocoids are effective for symptomatic relief and suppressing disease activity, although concerns over side effects limit their use. Commonly administered parenterally or as intra-articular injections, they should be reserved for use in acute disease flares or while waiting for DMARDs to take effect. Oral or pulsed intravenous therapy is effective for systemic manifestations of RA.
Surgical Management
Synovectomy, realignment and repair of tendons, joint prostheses and arthrodesis may be required for severe pain or deformity.
Prognosis
Although DMARDs and TNFα inhibitors may induce remission, for most patients RA is a progressive disorder, with up to 10% of cases suffering severe disability. Cardiovascular disease, infection and secondary amyloidosis are major causes of morbidity and mortality. Young age at onset, severe disease/disability at presentation, extra-articular manifestations and high RF titres all predict a worse prognosis.
Seronegative Spondyloarthropathies
A group of rheumatological disorders sharing common features:
- sacroileitis
- peripheral arthritis (typically large joints, especially of the lower limbs)
- mucocutaneous inflammation
- familial aggregation
- absence of rheumatoid factor (hence ‘seronegative’).
Psoriatic Arthritis
Psoriatic arthritis affects between 0.01 and 0.1% of the population, with a mean age of onset between 30 and 50 years and equal sex distribution. It is associated with HLA-DR4 (peripheral joint involvement) and HLA-B27 (spinal disease) haplotypes, and is more prevalent in HIV-positive subjects. A similar pathological process (increased vascularity with an inflammatory cell infiltrate) occurs in the joints as in the skin.
Clinical Presentation
Approximately 10% of patients with psoriasis develop arthritis. There is no correlation between the presence or severity of psoriatic skin changes and joint involvement. Several different (not mutually exclusive) patterns are recognised:
- Asymmetric oligoarthritis (
 30–50% of cases) typically affecting a few large or small joints. Diffuse swelling of the digits (dactylitis), in which one or two digits take on a ‘sausage-like’ appearance, is a distinctive feature.
30–50% of cases) typically affecting a few large or small joints. Diffuse swelling of the digits (dactylitis), in which one or two digits take on a ‘sausage-like’ appearance, is a distinctive feature.
- Symmetrical polyarthritis (
 20–40%).
20–40%).
- Sacroileitis and spondylitis (
 5–30%) which may be associated with Achilles tendonitis and plantar fasciitis.
5–30%) which may be associated with Achilles tendonitis and plantar fasciitis.
- Distal interphalangeal joint involvement (
 5–15%).
5–15%).
- Arthritis mutilans (up to 15%) causes gross joint destruction in which resorption of terminal digits and juxta-articular bone results in ‘telescoping’ of the digits.
Nail pitting and onycholysis may be the only evidence of underlying psoriasis, but a careful search for skin changes (including the scalp, hairline and behind the ears) should be performed. Pustular psoriasis of the palms and soles may be confused with the rash of Reiter syndrome (keratoderma blennorrhagica).
Investigation
There is no single diagnostic test for psoriatic arthritis and a high index of clinical suspicion is required. The following may be useful:
- normochromic, normocytic anaemia and raised inflammatory markers
- negative RF
- HLA-B27 positivity (
 20% of all cases, and 50% of cases with spondylitis)
20% of all cases, and 50% of cases with spondylitis)
- radiological evidence of sacroileitis, distal interphalangeal joint involvement or digital juxta-articular bone resorption.
Management
- Supportive measures – physiotherapy, aids.
- Simple analgesics.
- NSAIDs.
- DMARDs – especially for the symmetrical RA-like pattern, with methotrexate being the preferred option. Hydroxychloroquine should be avoided as it may cause psoriatic flares.
- Tumour necrosis factor-α inhibitors (anti-TNFα therapies) – current NICE guidance recommends considering etanercept, adalimumab and infliximab in those patients with ≥ three tender and swollen joints failing treatment with ≥ two DMARDs.
Prognosis
This is dependent on the pattern of disease. The symmetrical polyarthritis form follows a similar course to RA, sacroileitis/spondylitis resembles ankylosing spondylitis and oligoarticular disease tends to run a more benign course. Arthritis mutilans is associated with considerable disability.
Ankylosing spondylitis
Ankylosing spondylitis (AS) affects between 0.1% and 1% of white populations. It is strongly associated with HLA-B27 and affects males more commonly than females (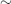 4 : 1), with a peak age of onset in adolescence/young adulthood. There may be a history of inflammatory bowel disease, psoriasis or reactive arthritis.
4 : 1), with a peak age of onset in adolescence/young adulthood. There may be a history of inflammatory bowel disease, psoriasis or reactive arthritis.
Enthesitis (inflammation of ligament or muscle tendon attachments to bone) is the cardinal pathological finding. Inflammation of the sacroiliac, facet and intervertebral joints is followed by ossification of spinal ligaments and intervertebral discs. Bony outgrowths from the vertebral margins extend vertically and coalesce. Eventually spinal fusion occurs.
Clinical Presentation
Spinal Symptoms
- pain (worse at night and in the morning, improving with exercise)
- stiffness (particularly after inactivity)
- reduced movement (especially the lumbosacral and cervical spine)
Advanced AS may result in a characteristic posture with cervical hyperextension, exaggerated thoracic kyphosis, loss of lumbar lordosis and compensatory knee flexion.
Systemic Symptoms
- large joint involvement (lower limbs)
- plantar fasciitis
- achilles tendinitis
- anterior uveitis
- apical pulmonary fibrosis ± respiratory failure (fixed ribcage with kyphoscoliosis)
- aortitis with aortic incompetence
- amyloidosis
Investigation
Diagnosis rests on the history and examination findings combined with the following.
Blood Tests
- rheumatoid factor negative
- ESR elevated (
 80% of cases) and CRP
80% of cases) and CRP
- HLA-B27 positivity (in 95% compared with 5–10% of the general population and 50% of asymptomatic relatives)
Radiography: Plain X-Ray Findings
- sacroileitis
- squaring of vertebrae
- syndesmophytes (bridging spurs of bone at the corners of adjacent vertebral bodies)
- facet joint involvement
- ossification (‘bamboo spine’)
Management
- Physiotherapy.
- NSAIDs.
- Sulphasalazine may be effective for peripheral joint involvement.
- Tumour necrosis factor-α inhibitors (anti-TNFα therapies): current NICE guidance recommends considering etanercept or adalimumab in patients with severe AS with evidence of sustained active spinal disease and where treatment with two or more NSAIDs for 4 weeks has failed to control symptoms.
- Corticosteroids are occasionally required.
Prognosis
With expert care most individuals will maintain complete or almost complete activity. In patients with more severe AS, moderate to severe bony ankylosis of the spine produces fixation of mobility and rounded kyphosis of the cervical and thoracic spine which may impair ventilation. In severe cases extreme rigidity of the spine may occur within 3–5 years. The disease may remit at any stage but recurrent episodes can occur. Poor prognostic indicators include onset in adolescence, high CRP and extraspinal joint involvement.
Reactive Arthritis (Reiter Syndrome)
The term reactive arthritis is used to denote joint inflammation arising in relationship to an infectious episode, which has usually resolved. The infective organism is not found within the joint itself as the inflammatory process probably results from an immune response to one or more bacterial antigens, following which activated T-lymphocytes and macrophages migrate to the synovium. Reactive arthritis is seen most commonly in young adults of either sex following infection with one of the following organisms:
- Chlamydia trachomatis
- Salmonella species
- Shigella species
- Campylobacter jejuni
- Yersinia enterocolitica
There is an increased incidence in populations where HLA-B27 is prevalent (HLA-B27 is involved in the presentation of bacterial antigens to CD8+ T cells).
Clinical Presentation
An episode of diarrhoea or urethritis may precede the onset of arthritis by up to a month. In up to half of all cases no prior infective episode can be identified. Clinical features include:
- Arthritis: acute or subacute, usually oligoarticular and asymmetrical affecting large joints of the legs (especially the knees). There may be associated fever and weight loss.
- Sacroiliitis: in up to 30% of cases.
- Plantar fasciitis and Achilles tendinitis.
- Conjunctivitis: common in the acute phase.
- Anterior uveitis is a feature of chronic recurrent disease, particularly when associated with sacroiliitis.
- Urethritis and circinate balanitis may persist in some patients.
- Pustular hyperkeratotic lesions of the soles of the feet and palms of the hands (keratoderma blennorrhagica) occurs in
 15% of patients.
15% of patients.
- Distal interphalangeal joint swelling or dactylitis may be seen in chronic disease.
Investigation
There is no single diagnostic test for reactive arthritis and a high index of clinical suspicion is required. The following may be useful:
- Raised inflammatory markers.
- Negative screen for IgM RF.
- Joint aspiration: fluid is turbid, but contains no organisms or crystals.
- HLA-B27 positive.
- Radiological changes: joint erosions and sacroiliitis may be seen.
- All patients should be screened for Chlamydia trachomatis infection, which can be clinically silent.
Management
Acute Phase
- simple analgesics
- NSAIDs
- joint aspiration and intra-articular injection of corticosteroids
Chronic Peripheral Joint Disease
- DMARDs (e.g. sulphasalazine, methotrexate) may be required.
- Treat underlying sexually transmitted infection (this does not influence the course of joint disease).
Prognosis
Most patients recover within weeks or months. A small number may suffer recurrence at a later date. For 15–30% it becomes a chronic disorder requiring on-going treatment. Persistence and recurrence are more likely in HLA-B27 positive individuals.
Enteric Arthropathy
Around 20% of patients with inflammatory bowel disease (Crohn’s disease, ulcerative colitis) develop an arthropathy, in the form of a peripheral mono- or oligoarticular arthritis or sacroiliitis. Treatment is with simple analgesics, NSAIDS, intra-articular or oral corticosteroids and, where necessary, DMARDs.
Autoimmune Rheumatic Disorders (Connective Tissue Diseases)
Systemic Lupus Erythematosus (SLE)
SLE is nine times more common in women than men and usually presents at age 20–40 years (90% of cases). It is exacerbated by exposure to ultraviolet radiation, infections, certain drugs, stress and pregnancy. In North America and Northern Europe the prevalence per 100,000 is estimated at 30–50 for white women, 100 for Asian women and 100–200 for African-Caribbean women. Cause unknown, it seems likely that environmental triggers act together with a genetic predisposition to cause the disease. HLA-B8, DR2 and DR3 are associated with SLE, and other non-HLA loci have been implicated.
The development of antinuclear antibodies (ANA positivity) is the key serological finding in patients with SLE, commonly with lymphocytic infiltration and deposition of immunoglobulins and immune complexes in affected tissues/organs, although whether antibodies drive the disease is uncertain. Vasculitis leads to ischaemic damage.
Clinical Presentation
Early manifestations are:
- fever
- arthralgia
- general ill health and fatigue
- weight loss
- skin rash.
SLE can mimic rheumatoid arthritis or bacterial endocarditis and may cause nephrotic syndrome. Major organ involvement may be present at the outset or can evolve over time. One or more of the following systems are typically involved (Fig. 18.2).
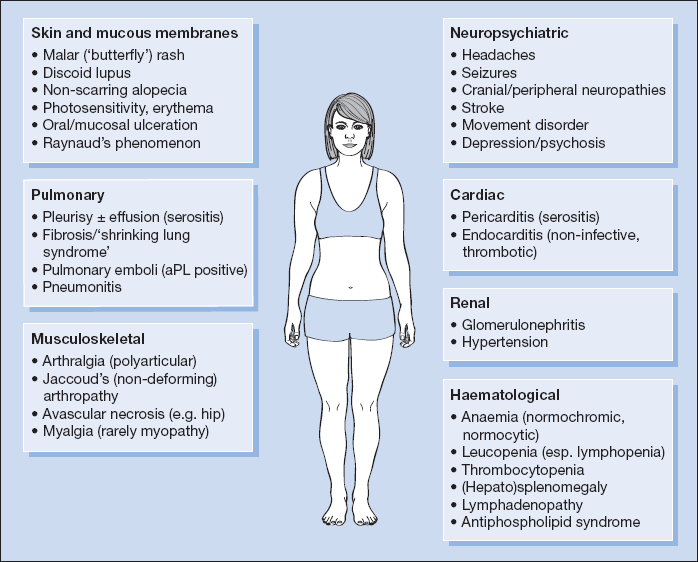
Musculoskeletal System (in 90% of Cases)
- Myalgia.
- Migratory polyarthralgia with early morning stiffness is common.
- Jaccoud’s arthropathy: non-deforming arthropathy caused by tendonitis rather than synovitis affecting the fingers, wrists, elbows, shoulders, knees and ankles.
- Avascular necrosis may follow prolonged corticosteroid therapy.
Skin and Mucous Membranes (in 80% of Cases)
Lupus may be confined to the skin as discoid or subacute cutaneous lupus; typically a raised, scarring rash on the face, scalp or limbs.
Other features include:
- Raynaud’s phenomenon in between a quarter and half of all cases
- non-specific erythema
- photosensitivity
- alopecia
- malar ‘butterfly’ rash – bridging the nose and cheeks in 30%
- oral and mucosal ulceration (30%)
- nail fold infarcts (10%)
- livedo reticularis
- panniculitis
- bullous eruptions
Kidneys (in 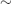 100% of Cases)
100% of Cases)
- SLE is associated with a range of glomerulonephritides. Almost all patients with SLE have histological abnormalities on renal biopsy and 50% develop overt renal involvement. When present, it is associated with a worse prognosis. Clinical presentation includes:
- hypertension
- haematuria
- proteinuria
- nephrotic syndrome
- acute kidney injury
- end-stage renal disease.
Neuropsychiatric Manifestations (in 50–60% of Cases)
Neurological involvement in lupus usually arises in the context of active systemic disease. Manifestations include:
- headache
- peripheral neuropathy
- cranial nerve abnormalities
- mononeuritis multiplex
- tremor
- strokes
- seizures
- psychoses/depression
- limb weakness/numbness.
Central nervous system (CNS) abnormalities are associated with a poorer prognosis.
Lungs (in 40–50% of Cases)
Commonly:
- pleurisy, occasionally with effusion
- patchy consolidation and areas of collapse
- diffuse reticulonodular shadowing on chest X-ray.
Rarely:
- ‘shrinking lung syndrome’
- lupus pneumonitis, which may be haemorrhagic, is rare but often fatal
- pulmonary emboli in patients who are antiphospholipid antibody positive.
Cardiovascular System (in 40% of Cases)
- mild pericarditis: may be the first presenting feature of SLE
- non-infective thrombotic endocarditis (Libman–Sacks)
- hypertension is usually associated with renal involvement
Haematology
- raised ESR in active disease
- normochromic normocytic anaemia
- mild lymphopenia
- mild thrombocytopenia
- occasionally severe thrombocytopenia, leucopenia and haemolytic anaemia
- antiphospholipid syndrome (
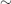 20%)
20%)
- reactive lymphadenopathy (30–40%)
- splenomegaly (10%)
The American College of Rheumatology has proposed criteria for the diagnosis of SLE based on a combination of clinical and laboratory features (Box 18.3).
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


