Fig. 6.1
Dose-dependent plasma concentrations of amisulpride in 179 schizophrenic patients treated and observed under everyday conditions. Drug concentrations are highly variable. Under recommended doses of 400–800 mg/day, many patients exhibit drug concentrations above or below the recommended therapeutic reference range of 100–320 ng/mL (Hiemke et al. 2011)
Similar patterns of dose-related drug concentrations are known for any psychotropic drug, e.g. antidepressants (Ostad Haji et al. 2012) or antipsychotic drugs (Hiemke et al. 2004; Hefner et al. 2013). Since the drug concentration in the blood usually correlates well with concentrations at the site of action, it has been proven for many drugs to use blood level measurements to guide pharmacotherapy, i.e. to use therapeutic drug monitoring (TDM). Using TDM, the dose should be corrected individually as shown in Fig. 6.2 when the drug concentrations in the blood are not within the therapeutic reference range and the patient did not improve or suffers from side effects. At concentrations within the therapeutic range, the highest probability of response and good tolerability is expected. In psychiatry, both too low and too high drug doses or concentrations are associated with risks. Too low doses or drug concentrations can be associated with treatment failure or relapse which bear morbogenic risks such as psychotic exacerbation or self-harming. Too high doses or drug concentrations can lead to adverse drug reactions or intoxications.
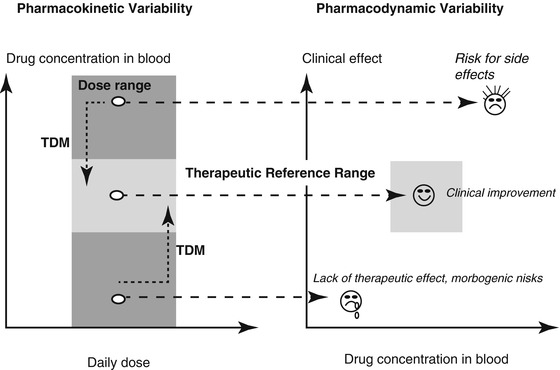
Fig. 6.2
The concept of therapeutic drug monitoring (TDM) for guidance of pharmacotherapy. Under recommended doses and steady concentration, drug in the blood should be within the therapeutic reference range to attain highest probability of drug response and good tolerability. In psychiatry, too low or too high drug concentrations are associated with risks. Too low doses/concentrations bear morbogenic risks due to treatment failure or relapse like psychotic exacerbation or self-harming. Too high drug concentrations can lead to reduced tolerance and poor compliance or adverse drug reactions
TDM consists not only of determination and reporting of drug concentrations in blood. It is a process that begins with the indication for TDM and ends with the treatment decision for the patient. TDM was originally established for drugs with a narrow therapeutic index, such as digoxin or cyclosporin A, and in psychiatry, for lithium (Fry and Marks 1971), tricyclic antidepressants (Åsberg et al. 1971; Preskorn and Fast 1991) or the antipsychotic clozapine (Simpson and Cooper 1978). Primary aims of TDM in psychiatry were originally related to safety, drug defaulting and side effects (Preskorn and Fast 1992; Sjöqvist et al. 1980; Touw et al. 2005).
Since the first application of TDM between 1970 and 1980, enormous progress was made in psychopharmacotherapy not only by the development of new drugs but also by many new findings on the pharmacokinetics and pharmacodynamics of available drugs. Cytochromes P450 (CYP) enzymes have been recognized as a major source of variability in drug pharmacokinetics and response (Zanger et al. 2014). Of 57 putatively functional human CYP enzymes, seven were found to be relevant for about 90 % of phase I metabolism of psychoactive drugs, CYP1A2, CYP2B6, CYP2C9, CYP2C19, CYP2D6, CYP2E1 and CYP3A4. The expression of each CYP is influenced by multiple mechanisms and factors including genetic polymorphisms, induction by xenobiotics, regulation by cytokines and hormones and alterations during disease states, as well as sex, age and others (Zanger and Schwab 2013). Genetic polymorphisms play a major role for the function of CYP2D6 and CYP2C19 and lead to distinct pharmacogenetic phenotypes termed poor (PM, no active alleles), intermediate (IM), extensive (EM, basic genotype) and ultrarapid metabolizers (UM).
Co-medication and other xenobiotics are another important determinant that has been found to affect the pharmacokinetics of drugs. Fluoxetine, fluvoxamine, paroxetine, bupropion, duloxetine, moclobemide and multiple other drugs were identified as potent inhibitors of distinct CYP enzymes. To a lesser degree, but also clinically relevant, carbamazepine, compounds in St. John’s wort and other drugs were found to enhance the expression of CYP3A4. Marked pharmacokinetic drug-drug interactions can result when combining such inhibitors or inducers with drugs that are metabolized by inhibited or induced enzymes (victim drugs). The escalating use of prescribed drugs has led to polypharmacy as a “normal” condition of pharmacotherapy, in psychiatry as well as in other disciplines (Guthrie et al. 2015), especially in elderly patients.
Modern TDM uses the above-mentioned psychopharmacology knowledge gained during the last 40 years in practice for the best possible pharmacotherapy of individual patients. It thus enhances patient care and patient safety. Modern pharmacovigilance, which is defined as the science and activities relating to the detection, assessment, understanding and prevention of adverse effects or any other drug-related problem (WHO 2002; EMA 2014), also aims to enhance patient care and patient safety in relation to the use of medicines. Spontaneous reporting of suspected adverse drug reactions (ADRs) has long been the cornerstone of pharmacovigilance worldwide for the identification of early signals of problems of safety related to the use of medicines. However, data generated from spontaneous reporting systems are often fragmentary. They are of limited use for the evaluation of observed events. Spontaneously reported adverse drug reactions are therefore considered as a hint that must be followed by systematic studies. Since TDM supervises pharmacotherapy under everyday conditions and thus aims to prevent safety problems related to the use of medicines (Haen 2011; Jaquenoud-Sirot et al. 2006), TDM and pharmacovigilance thus have the same primary aims. When applied appropriately, i.e. as recommended in the consensus guidelines for TDM in psychiatry, collected information is more complete and better structured (Baumann et al. 2004; Hiemke et al. 2011) than reports in the pharmacovigilance programs. TDM is therefore potentially a highly suitable tool and source for pharmacovigilance.
6.2 Monitoring of Plasma Concentrations to Guide and Supervise Psychopharmacotherapy
The benefits of monitoring of plasma concentrations regarding the optimization of pharmacotherapy can only be obtained if the method is adequately integrated into the clinical treatment process. The TDM group of the Arbeitsgemeinschaft für Neuropsychopharmakologie und Pharmakopsychiatrie (AGNP) issued best practice guidelines for TDM in psychiatry in 2004 (Baumann et al. 2004) which were updated in 2011 (Hiemke et al. 2011).
6.2.1 Therapeutic Reference Range and Laboratory Alert Level
TDM is based on the assumption that there is a relationship between plasma concentrations and clinical effects, i.e. therapeutic improvement, side effects and adverse effects (Hefner et al. 2013). It also assumes that there is a plasma concentration range of the drug which is characterized by maximal effectiveness and maximal safety, the “therapeutic reference ranges” (often called “therapeutic window”). The therapeutic reference range is a key target value to perform TDM.
The therapeutic reference ranges reported in the AGNP guideline define ranges of medication concentrations which specify a lower limit below which a drug-induced therapeutic response is relatively unlikely to occur and an upper limit above which tolerability decreases or above which it is relatively unlikely that therapeutic improvement may be still enhanced. It is an orienting, population-based range which may not necessarily be applicable to all patients. Individual patients, e.g. patients under drug combinations, may show optimal therapeutic response under a drug concentration that differs from the therapeutic reference range. The therapeutic reference ranges as recommended by the TDM group of the AGNP for antidepressant, antipsychotic and mood-stabilizing drugs are given in Table 6.1. They were evidence based and derived from the literature by a structured review process. For only 15 neuropsychiatric drugs, therapeutic reference ranges based on randomized clinical trials were found in the literature. For most drugs, reference ranges were obtained from studies with therapeutically effective doses.
Table 6.1
Therapeutic reference ranges and laboratory alert levels of antidepressant, antipsychotic and mood-stabilizing drugs in blood plasma or serum used for therapeutic drug monitoring (TDM)-guided pharmacotherapy
Drug and active metabolite | Therapeutic reference range | Laboratory alert level | Comment |
|---|---|---|---|
Antidepressant drugs | |||
Amitriptyline plus nortriptyline | 80–200 ng/mL | 300 ng/mL | Elevated concentration in PM of CYP2D6 |
Bupropion plus hydroxybupropion | 225–1,500 ng/mL | 2,000 ng/mL | Bupropion is unstable at room temperature; reference range and alert level refer to hydroxybupropion only |
Citalopram | 50–110 ng/mL | 220 ng/mL | Elevated concentration in PM of CYP2C19 |
Clomipramine plus norclomipramine | 230–450 ng/mL | 450 ng/mL | Elevated concentration in PM of CYP2D6 and CYP2C19 |
Desipramine | 100–300 ng/mL | 300 ng/mL | Elevated concentration in PM of CYP2D6 |
Desvenlafaxine | 100–400 ng/mL | 600 ng/mL | Elevated concentration in PM of CYP2C19 |
Doxepin plus nordoxepin | 50–150 ng/mL | 300 ng/mL | Elevated concentration in PM of CYP2D6 or CYP2C19 |
Duloxetine | 30–120 ng/mL | 240 ng/mL | Lower concentration in smokers than in nonsmokers, moderate inhibitor of CYP2D6 |
Escitalopram | 15–80 ng/mL | 160 ng/mL | Higher concentration in PM of CYP2C19 |
Fluoxetine plus norfluoxetine | 120–500 ng/mL | 1,000 ng/mL | Long elimination half-life of norfluoxetine (mean 14 days) and long-lasting inhibition of CYP2D6 |
Fluvoxamine | 60–230 ng/mL | 500 ng/mL | Inhibitor of CYP1A2 and CYP2C19 |
Imipramine plus desipramine | 175–300 ng/mL | 300 ng/mL | Elevated concentration in PM of CYP2D6 |
Maprotiline | 75–130 ng/mL | 220 ng/mL | Active metabolite N-desmethylmaprotiline |
Mianserine | 15–70 ng/mL | 140 ng/mL | |
Milnacipran | 50–110 ng/mL | 220 ng/mL | No phase I metabolism |
Mirtazapine | 30–80 ng/mL | 160 ng/mL | |
Moclobemide | 300–1,000 ng/mL | 2,000 ng/mL | Inhibitor of CYP2C19 and CYP2D6 |
Nortriptyline | 70–170 ng/mL | 300 ng/mL | Elevated concentration in PM of CYP2D6 |
Paroxetine | 30–60 ng/mL | 120 ng/mL | Inhibitor of CYP2D6 |
Reboxetine | 60–350 ng/mL | 700 ng/mL | |
Sertraline | 10–150 ng/mL | 300 ng/mL | |
Tianeptin | 30–80 ng/mL | 160 ng/mL | |
Tranylcypromin | ≤50 ng/mL | 100 ng/mL | Irreversible inhibition of monoamine oxidase (MAO); drug actions correlate with MAO activity but not with drug concentration in blood |
Trazodone | 700–1,000 ng/mL | 1,200 ng/mL | |
Trimipramine | 150–300 ng/mL | 600 ng/mL | Active metabolites |
Venlafaxine plus O-desmethylvenlafaxine | 100–400 ng/mL | 800 ng/mL | Elevated concentration of venlafaxine in PM of CYP2D6 or CYP2C19 |
Vortioxetine | 10–60 ng/mL | 120 ng/mL | Elevated concentration in PM of CYP2D6 |
Antipsychotic drugs | |||
Amisulpride | 100–320 ng/mL | 640 ng/mL | |
Aripiprazole | 150–500 ng/mL | 1,000 ng/mL | Active metabolite dehydroaripiprazole |
Asenapine | 2–5 ng/mL | 10 ng/mL | |
Benperidol | 1–10 ng/mL | 20 ng/mL | Higher levels may be required in patients under long-term high-dose therapy due to adaptive changes |
Bromperidol | 12–15 ng/mL | 30 ng/mL | |
Chlorpromazine | 30–300 ng/mL | 600 ng/mL | |
Chlorprothixene | 20–300 ng/mL | 400 ng/mL | |
Clozapine | 350–600 ng/mL | 1,000 ng/mL | Major metabolite N-desmethylclozapine |
Flupenthixol | 0.5–5 ng/mL (cis-Isomer) | 15 ng/mL | |
Fluphenazine | 1–10 ng/mL | 15 ng/mL | |
Fluspirilen | 0.1–2.2 ng/mL | 4.4 ng/mL | |
Haloperidol | 1–10 ng/mL | 15 ng/mL | Higher levels can be tolerated in patients under long-term high-dose therapy due to adaptive changes |
Iloperidone | 5–10 ng/ml | 20 ng/ml | |
Levomepromazine | 30–160 ng/mL | 320 ng/mL | Inhibitor of CYP2D6 |
Lurasidone | 40–120 ng/mL | 240 ng/mL | |
Melperone | 30–100 ng/mL | 200 ng/mL | Inhibitor of CYP2D6 |
Olanzapine | 20–80 ng/mL | 100 ng/mL | Under olanzapine pamoate, patients exhibited a postinjection syndrome when drug concentrations exceeded 100 ng/mL |
Paliperidone | 20–60 ng/mL | 120 ng/mL | Paliperidone = 9-hydroxyrisperidone |
Perazine | 100–230 ng/mL | 460 ng/mL | |
Perphenazine | 0.6–2.4 ng/mL | 5 ng/mL | |
Pimozide | 15–20 ng/mL | 20 ng/mL | |
Pipamperone | 100–400 ng/mL | 500 ng/mL | |
Prothipendyl | 5–10 ng/mL | 20 ng/mL | |
Quetiapine | 100–500 ng/mL | 1,000 ng/mL | When the patient has taken the extended release (XR) formulation in the evening and blood was withdrawn in the morning, plasma concentrations are by mean twofold higher than trough levels |
Risperidone plus 9-hydroxyrisperidone | 20–60 ng/mL | 120 ng/mL | Elevated concentration of risperidone in PM of CYP2D6 |
Sertindole | 50–100 ng/mL | 200 ng/mL | Active metabolite dehydrosertindole (concentration at therapeutic doses 40–60 ng/mL) |
Sulpiride | 200–1,000 ng/mL | 1,000 ng/mL | Renal elimination |
Thioridazine | 100–200 ng/mL | 400 ng/mL | Contraindicated in poor metabolizers of CYP2D6, elevated concentration |
Ziprasidone | 50–200 ng/mL | 400 ng/mL | The drug should be taken with a meal; otherwise absorption is reduced and plasma concentration will be lower than expected |
Zotepine | 10–150 ng/mL | 300 ng/mL | |
Zuclopentixol | 4–50 ng/mL | 100 ng/mL | |
Mood-stabilizing drugs | |||
Carbamazepine | 4–10 μg/mL | 20 μg/mL | Active 10,11-epoxide metabolite contributes to clinical effects |
Lamotrigine | 3–14 μg/mL | 30 μg/mL | So far no specific reference range for mood-stabilizing effect; valproate increases elimination half-life to 48–70 h |
Lithium | 0.5–1.2 mmol/l | 1.2 mmol/l | Renal elimination |
(4–8 μg/mL) | (8 μg/mL) | ||
Valproic acid | 50–100 μg/mL | 120 μg/mL | In individual cases, 120 μg/mL is also tolerated in acute mania |
If prospective or retrospective studies on the therapeutic reference range are lacking, the drug concentration that is expected under steady-state conditions (Css) can be calculated for the given dose as follows:
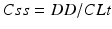 The calculation is based on the direct correlation of the daily dose (DD, constant dose per day under steady state) to its blood concentration Css, with the total clearance of the drug (CLt) being the correlation coefficient:
The calculation is based on the direct correlation of the daily dose (DD, constant dose per day under steady state) to its blood concentration Css, with the total clearance of the drug (CLt) being the correlation coefficient:
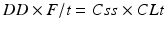 with DD as dose (mg), F as bioavailability and τ as dosing interval (h). Based on this information, it is possible to calculate the plasma concentration of a drug that may be expected in blood under a defined dose (Haen et al. 2008).
with DD as dose (mg), F as bioavailability and τ as dosing interval (h). Based on this information, it is possible to calculate the plasma concentration of a drug that may be expected in blood under a defined dose (Haen et al. 2008).
In addition to the therapeutic reference range and especially with regard to safety aspects, the so-called laboratory alert level is another relevant target value for TDM-guided drug therapies. For most psychotropic drugs, plasma concentrations with an increased risk of toxicity are normally much higher than the upper threshold levels of the therapeutic reference ranges shown in Table 6.1. In the TDM guidelines (Hiemke et al. 2011), a “laboratory alert level” mostly above the upper plasma concentration limit of the therapeutic reference range was defined as follows: “The “laboratory alert levels” (Table 6.1) indicate drug concentrations that cause the laboratory to feedback immediately to the prescribing physician. The alert levels are based on reports on intolerance or intoxications and plasma concentration measurements. In most cases, however, it was arbitrarily defined as a plasma concentration that is twofold higher than the upper limit of the therapeutic reference range. The laboratory alert should lead to dose reduction when the patient exhibits signs of intolerance or toxicity. When the high drug concentration is well tolerated by the patient and if dose reduction bears the risk of symptom exacerbation, the dose should remain unchanged. The clinical decision, especially in case of unchanged dose, needs to be documented in the medical file.
6.2.2 TDM Request
TDM should only be requested when there is evidence that the result will provide an answer to a specific question. Typical indications are shown in Fig. 6.3.
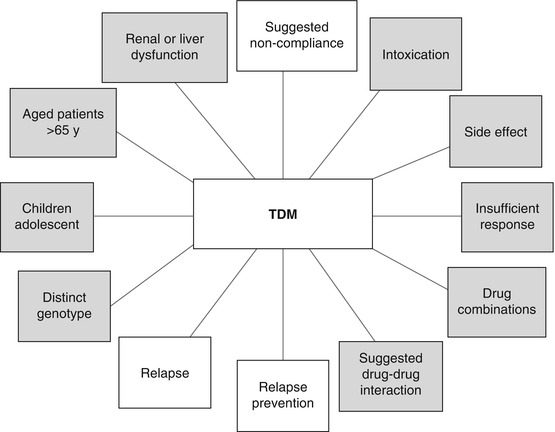
Fig. 6.3
Typical indications to request therapeutic drug monitoring (TDM). Indications related to pharmacovigilance are marked in grey
TDM requests must include a completed request form, especially when information is to be used for pharmacovigilance issues. An adequate interpretation of the results is essential to support clinical decision making. The form should contain the following:
Patient name or code
Demographic data
Diagnosis
Medication including co-medications
Reason for the request
Commercial and the generic name of the drug and its dose
Galenic formulation
Time of the last change of the dose
Time of blood withdrawal
Moreover,
A brief clinical rating score for evaluation of therapeutic improvement
A brief side effect rating score
should be on the request form (Hiemke et al. 2011).
For objective symptom rating, the clinical global impression (CGI) scale, which measures severity of illness and therapeutic improvement (Guy 1976), is useful. For safety purposes, the summary form of the UKU scale (Lingjærde et al. 1987) is useful to evaluate the occurrence and severity of side effects.
A brief comment on the clinical situation must also be given for interpretation of the results.
6.2.3 Blood Sample Collection, Storage and Shipment
Generally, TDM is carried out in plasma or serum samples. There is no consensus whether plasma or serum should be preferred. The few available comparisons indicate that values obtained from serum or plasma can be used interchangeably. Analysis of drugs in other materials such as urine, spinal fluid, tears, hairs or maternal milk has not been introduced for TDM purposes, and no validated data are available which deal with therapeutic concentrations. Saliva offers the advantage of non-invasive collection.
With few exceptions, TDM relies on trough steady-state plasma concentrations. One exception is the indication “side effects”. When they occur, it is useful to know if they can be attributed to elevated drug concentration and if the dose may be reduced without loss of efficacy. For concentration measurements under steady state, blood should be collected after four to five drug elimination half-lives after the start of or change of dosage. For most psychotropic drugs, elimination half-lives vary between 12 and 36 h. Notable exceptions are quetiapine, trazodone or venlafaxine, which display elimination half-lives around 6 h. Fluoxetine and aripiprazole have longer elimination half-lives. In clinical practice, the appropriate sampling time for most psychoactive drugs is 1 week after stable daily dosing and immediately before ingestion of the morning dose, which usually is 12–16 h (or 24 h if the drug is given once daily in the morning) after the last medication. It is always recommended to indicate exactly the time of administration of the last dose for interpretation.
With few exceptions, serum or plasma samples can be stored in the dark (at 4 °C) for at least 24 h, and most drug samples can be sent without freezing. An exception is bupropion. Blood serum or plasma must be frozen after blood withdrawal. Olanzapine must also be stored frozen (−20 °C) if not analysed within 72 h.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


