Fig. 35.1
Examples of unnatural peptidomimetics
As they are not based on regular peptide backbones, they are often much more stable against proteolysis and have improved bioavailability compared to peptide counterparts [29]. In addition, peptidomimetics greatly enhance structure diversities by not only introducing novel backbones, but also displaying diverse side functional groups with virtually limitless potential. Although peptidomimetics were initially designed to mimic the primary structures of peptides, most classes of these peptide mimics are also able to fold into well-defined discrete three-dimensional structures such as sheets [30–32], helices [33], turns [34–36], and tertiary structures [37–44]. As such, they can mimic structures and functions of peptides and proteins and have begun to find potential applications in translational medicine [45, 46]. These classes of peptidomimetics have been extensively explored in the last two decades and reviewed comprehensively [10, 14, 29, 45, 47–50].
35.1.3 AApeptides
We have recently designed a new class of peptidomimetics termed “AApeptides” based on chiral PNA backbone [51–61]. They are termed “AApeptides” as they contain N–acylated-N–aminoethyl amino acid residues (Fig. 35.2). Depending on the positions of side chains on the backbone, two related classes of AApeptides—α-AApeptides and γ-AApeptides—were developed.
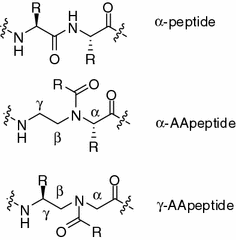
Fig. 35.2
The structures of α-peptides, α-AApeptides, and γ-AApeptides
Both α– and γ-AApeptides were designed to mimic the primary structures of canonical α-peptides so as to facilitate drug discovery. Each AApeptide residue is comparable to a dipeptide residue. Although they have different backbones compared to α-peptides, AApeptides project same number of side chains as regular peptides of same lengths do [51, 52]. In addition, AApeptides have limitless diversity in their functional groups, as half of side chains are introduced to the backbones through acylation by any carboxylic acids. Furthermore, due to their unnatural backbone, they are highly resistant to enzymatic degradation [51, 52]. More importantly, the synthesis of AApeptides is modular and straightforward, by utilizing solid-phase synthesis to assemble AApeptide building blocks together [51, 52, 62, 63]. As such, AApeptides can be developed to mimic the structures and functions of bioactive peptides. Indeed, certain AApeptide can bind to RNA with high affinity comparable to the Tat peptide [64], and some AApeptides are efficient cell-penetrating peptidomimetics which can translocate cellular membrane, suggesting their potential for drug delivery [65, 66]. AApeptides also have found great potential in material sciences as some of them are able to from novel nanorods and nanoparticles [67]. In this chapter, we will focus on their application as potential therapeutics for translational medicine, including their development as novel antimicrobial and anticancer agents.
35.2 Antimicrobial Agents
35.2.1 Background
One of the biggest clinical and public concerns worldwide is the antibiotic resistance [68], attributed to antibiotic abuse [69, 70]. The emergence of multi-drug-resistant bacterial strains such as methicillin-resistant Staphylococcus aureus (MRSA), vancomycin-resistant enterococci (VRE), and methicillin-resistant Staphylococcus epidermidis (MRSE) has largely abolish the efficacy of conventional antibiotics [71–73]. There has been tremendous effort in the search of new and effective antibiotics. Antimicrobial peptides (AMPs) are of significant interest for such development. AMPs exist virtually in all organisms and are their first line of defense [69, 70, 74–76]. Although the exact bactericidal mechanisms of AMPs may vary from one to the others [71], the hallmark of their structures is that they form globally amphipathic conformations upon interaction with bacterial membranes. In such amphiphilic structures, cationic and hydrophobic side groups segregate into discrete regions. The cationic region first associates with negatively charged bacterial membranes through electrostatic interaction [69, 70, 74–76], and then, the hydrophobic side chains disrupt membranes through hydrophobic interactions. Although such interactions lack defined membrane targets and rather biophysical, the selectivity of AMPs is high because mammalian cell membranes are largely zwitterionic. On the other hand, since antimicrobial activity of AMPs lacks defined bacterial internal and membrane targets, the probability of inducing antibiotic resistance is much lower. Additionally, the amphipathic structures of AMPs are not relevant to precise sequences and defined secondary structures, and in fact, the examples of AMPs embrace α-helices, β-sheets, and extended conformation. [70]
However, there are obstacles for the development of AMPs as new generation of antibiotics. These include susceptibility toward protease degradation, low-to-moderate antimicrobial activity, and limited diversity for optimization. Antimicrobial peptidomimetics, developed to mimic the function of AMPs, can potentially overcome these disadvantages. Many classes of antimicrobial peptidomimetics have been developed, including peptoids [33, 77–81], β-turn mimetics [82, 83], arylamide oligomers [71, 84, 85], and β-peptides [86–88], which have been comprehensively reviewed [69, 70, 89]. The development of these antimicrobial peptidomimetics has led to the recognition that defined secondary structures such as helical or sheet-like structures are not required for antimicrobial activity [62, 63, 90–92].
35.2.2 Design of Antimicrobial AApeptides
AApeptides are expected to be good candidates for the design of novel antimicrobial agents that mimic AMPs, as they possess backbones with limited conformational flexibility and [62, 90] high stability [51, 52, 93]. As such, amphipathic AApeptides can be designed in a very straightforward way by joining amphiphilic AApeptide building blocks, since AApeptides comprising of amphiphilic building blocks (containing one hydrophobic and one cationic side chain) are expected to adopt globally amphipathic structures upon interaction with bacterial membranes (Fig. 35.3). Their activity and selectivity can be further tuned through the introduction of additional hydrophobic (containing two hydrophobic side chains) building blocks, as well as changing the nature of the hydrophobic and cationic groups.
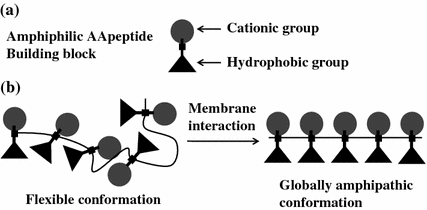
Fig. 35.3
Illustration of the design of antimicrobial AApeptide. a The structure of amphiphilic AApeptide building block. b The formation of amphipathic structure of AApeptides on bacterial membranes. Adapted from Ref. [90]
35.2.3 α-AApeptides as Antimicrobial Agents
35.2.3.1 Linear Antimicrobial α-AApeptides
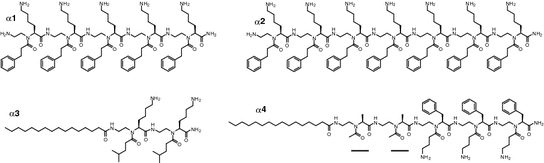
Following the design strategy, a small library of linear α-AApeptide sequences was prepared, which led to the identification of two lead sequences α 1 and α 2 (Fig. 35.4) [62]. These linear sequences contain amphiphilic α-AApeptide building blocks and were expected to adopt globally amphipathic structures upon interaction with bacterial membranes so as to kill bacterial pathogens. The amphiphilic sequence α 1 is active against Bacillus subtilis and Staphylococcus epidermidis, and addition of one more amphiphilic building block led to the identification of the more potent antimicrobial agent α 2 [62]. Fluorescence microscopy and TEM studies suggested that α 2 exert bactericidal activity by damaging bacterial membranes [62]. The preliminary data of linear α-AApeptides antimicrobial agents demonstrate that α-AApeptides are good candidates to mimic the structure and mechanism of AMPs. The design seems straightforward: Amphiphilic building-block-based α-AApeptides tend to selective kill bacteria pathogens, and longer sequences are more active as they contain more hydrophobic and cationic groups.
35.2.3.2 Lipo-Antimicrobial α-AApeptides
Some natural lipopeptide antibiotics have been used as antibiotics to treat bacterial pathogens [95], although they are different from host-defense peptides in their antimicrobial mechanisms. As they are produced as metabolites in bacteria and fungi [95, 96] and are either anionic [97], cationic [98], and neutral [99], many of these natural lipo-antibiotics are narrow spectrum in their activity. Although their antimicrobial mechanism may vary, lipid tail is generally believed to largely contribute to the antimicrobial activity. Therefore, lipo-sequences with small number of hydrophobic and cationic groups could be very active [100].
It is expected that cationic lipo-α-AApeptides should mimic AMPs because their overall structure is still cationic, and as such, they are active against both Gram-positive and Gram-negative bacteria. Indeed, with just two amphiphilic building blocks, lipidated α-AApeptide α 3 shows potent activity against Gram-positive bacteria [94]. The antimicrobial activity is further enhanced for α 4, which contains two more hydrophobic building blocks and one amphiphilic building block, possibly due to the stronger interaction with bacteria membranes [94].
35.2.4 γ-AApeptides as Antimicrobial Agents
35.2.4.1 Linear Antimicrobial γ-AApeptides
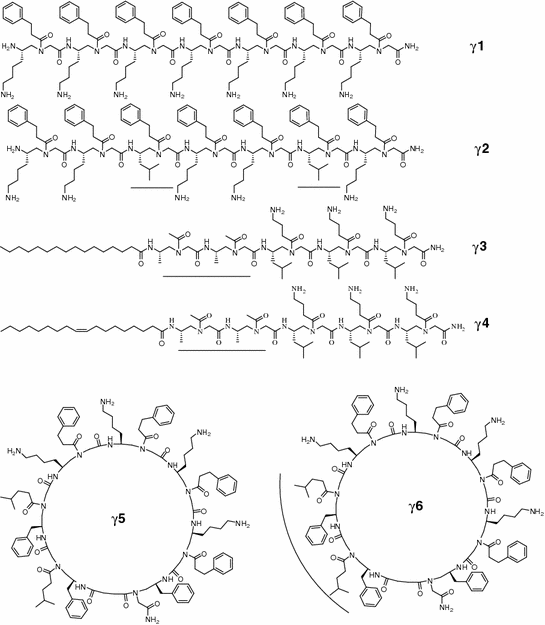
Similar design strategy was used to develop antimicrobial γ-AApeptides. Two most potent linear γ-AApeptides γ 1 and γ 2 are shown in Fig. 35.5 [90]. The amphiphilic sequence γ 1, containing seven amphiphilic building blocks, are active against several Gram-positive bacteria. Replacement of two amphiphilic building blocks in γ 1 with two hydrophobic building blocks led to the identification of γ 2 [90], which are broadly active against a range of Gram-positive and Gram-negative drug-resistant bacterial pathogens. Subsequent drug-resistant studies and fluorescent microscopy supported that γ 2 kill bacteria through membrane disruption [90].
35.2.4.2 Lipo-Antimicrobial γ-AApeptides
Cationic lipo-γ-AApeptides were also investigated for their antimicrobial activity. As shown in Fig. 35.5, both γ 3 and γ 4 have broad spectrum activity. The difference between γ 3 and γ 4 is that γ 4 has an unsaturated bond in its alkyl tail. Interestingly, γ 4 is more selective against bacteria than γ 3. Although mechanistic studies are required to elucidate the behavior, the findings might be significant for the development of new antimicrobial peptidomimetics. Further mechanistic studies including membrane depolarization, fluorescence microscopy, and drug-resistant assays suggested that lipo-γ-AApeptides can also mimic bactericidal mechanism of AMPs by killing bacterial pathogens through the disruption of bacterial membranes [63].
35.2.4.3 Cyclic Antimicrobial γ-AApeptides
Cyclic peptide antibiotics such as tyrocidine and protegrin I are common in nature. These cyclic AMPs have semi-rigid backbone conformations due to cyclization [83], which is believed to improve antimicrobial activity than linear sequences without any constraints, as they may possess more stable amphipathic structures. Similar strategy to design other classes of antimicrobial AApeptides was applied to generate focused cyclic γ-AApeptide library that mimics the mechanisms of AMPs [101]. This is achieved by cyclizing γ-AApeptide amphiphilic building blocks on the solid phase [101]. By varying the ratio of cationic/hydrophobic groups, their antimicrobial activity and selectivity can be fine-tuned [90]. Two cyclic γ-AApeptides γ 5 and γ 6 were identified from the library [101], and they show much improved antimicrobial activity against a range of multi-drug-resistant Gram-positive and Gram-negative bacterial pathogens than linear ones. Moreover, bearing more hydrophobic groups, γ 6 is found to be more potent than γ 5, suggesting that hydrophobic groups are important for the disruption of bacterial membranes.
35.3 Anticancer Agents
35.3.1 Disruption of MDM2-p53 Protein–Protein Interaction
The p53 protein is a tumor suppressor that integrates cellular stress signals and functions as a transcription factor with many target genes involved in DNA repair, cell cycle arrest, and activation of apoptosis. MDM2 (murine double minute two protein) is a p53-specific ubiquitin E3 ligase and thus promotes the proteasomal degradation of p53. It binds to the α-helical transactivation domain near the N-terminus of p53 and blocks the DNA-binding activity of p53 [102–104]. The disruption of p53/MDM2 protein–protein interactions can regulate apoptosis and therefore is of significant interest in the development of anticancer agents for translational medicine.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


